
Events
 Event
Materials Science
Event
Materials Science
- Jun 9th-10th, 2026
Frontiers in Digital Chemistry: Industry Summit
Schrödinger is pleased to host the inaugural Frontiers in Digital Chemistry: Industry Summit, an in-person user group meeting (UGM) dedicated to the digital chemistry community.
 Webinar
Life Science
Webinar
Life Science
- Jun 10th-16th, 2026
Expanding RetroSynth: Dynamic AI-driven synthesis planning proven to move your designs from bench to synthesis
In this webinar, we revisit RetroSynth, Schrödinger’s AI-powered solution for intelligent synthesis planning.
 Webinar
Life Science
Webinar
Life Science
- Jun 11, 2026
Generative Glide: AI-driven ultra-large virtual screening for real-world drug discovery
Join us as we go beyond slides and run a demo of the workflow, showing how Generative Glide performs in practice from setup through results.
Webinars
Webinar
Life Science
- Jun 10th-16th, 2026
Expanding RetroSynth: Dynamic AI-driven synthesis planning proven to move your designs from bench to synthesis
In this webinar, we revisit RetroSynth, Schrödinger’s AI-powered solution for intelligent synthesis planning.
Webinar
Life Science
- Jun 11, 2026
Generative Glide: AI-driven ultra-large virtual screening for real-world drug discovery
Join us as we go beyond slides and run a demo of the workflow, showing how Generative Glide performs in practice from setup through results.
 Webinar
Life Science
Webinar
Life Science
- Jun 24th – Jul 2nd, 2026
Modeling Services: A high-velocity entry point to the Schrödinger platform
Join us to learn how Schrödinger’s Modeling Services offer a fast-track gateway to the transformative power of our platform for your program.
Documentation
- Documentation
Learning Path: Oligonucleotide Modeling
A structured overview of tools and workflows for nucleic acids in drug discovery.
- Documentation
WaterMap
Efficiently converged MD simulations are run with explicit water molecules, and resultant trajectories are analyzed to cluster hydration sites.
- Documentation
SiteMap
Identify binding sites, including allosteric binding sites and protein-protein interfaces, and evaluate their druggability.
Tutorials
- Tutorial
Structure-Based Virtual Screening using Glide
Prepare receptor grids for docking, dock molecules and examine the docked poses.
- Tutorial
Ligand Binding Pose Generation for FEP+
Generate starting poses for FEP simulations for a series of BACE1 inhibitors using core constrained docking.
- Tutorial
Homology Modeling of Protein-Ligand Binding Sites with IFD-MD
Create a homology model of TYK2 from JAK3 and including a bound ligand. Compare this model with the crystal structure for TYK2 bound to 4GIH.
Training Videos
 Video
Life Science
Video
Life Science
Getting Going with Maestro BioLuminate
A free video series introducing the basics of using Maestro Bioluminate.
 Video
Life Science
Video
Life Science
- Video
Introducing Ligand Designer
An overview of the LigandDesigner workflow, Editing in 2D and 3D, using display options and overlays, and accessing the Admin Panel.
Publications
- Publication
- May 8, 2026
Discovery of 2H-Pyrrolo[3,4-c]pyridin-3-one Derivatives as Type-III c-MET Inhibitors Enabled by Free-Energy Perturbation CalculationsCl
Therrien, et al. ACS Medicinal Chemistry Letters, 2026
- Publication
- Apr 8, 2026
Structure-Based Discovery of Imidazo[4,5-c]pyridine SARM1 Modulators Showing Paradoxical Activation
Albanese, et al. Journal of Medicinal Chemistry, 2026, 69(8), 9521–9536
- Publication
- Mar 21, 2026
Structure-Based Calculation of Excipient Effects on the Viscosity of Concentrated Antibody Solutions
Shelley, et al. mAbs, 2026, 18(1)
Case Studies
 Case Study
Life Science
Materials Science
Case Study
Life Science
Materials Science
 Case Study
Life Science
Case Study
Life Science
 Case Study
Life Science
Case Study
Life Science
White Papers
 White Paper
Life Science
White Paper
Life Science
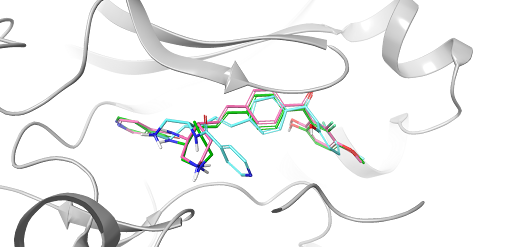
- Jan 29, 2026
FEP+ Pose Builder — maximizing utility and productivity in FEP simulations
FEP+ Pose Builder is a methodological advancement introduced as an integrated feature to drastically enhance accessibility, user-friendliness, and productivity within the FEP+ pipeline.
 White Paper
Life Science
White Paper
Life Science
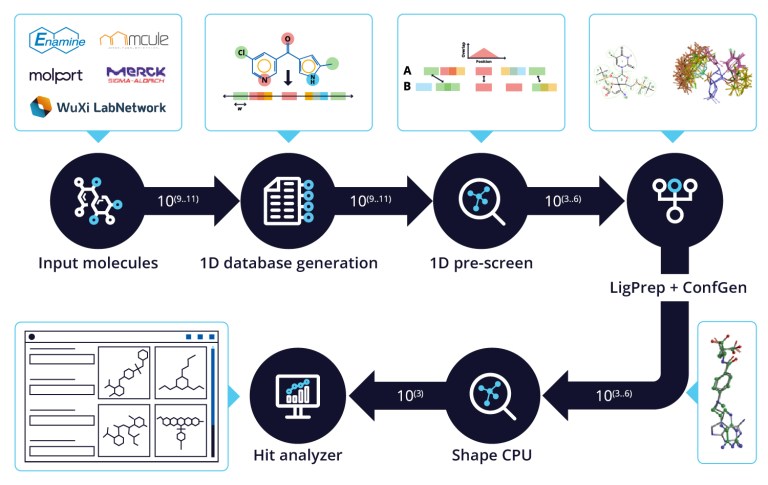
- Oct 29, 2024
20 Years of Glide: A Legacy of Docking Innovation and the Next Frontier with Glide WS
Glide has long set the gold standard for commercial molecular docking software due to its robust performance in both binding mode prediction and empirical scoring tasks, ease of use, and tight integration with Schrödinger’s Maestro interface and molecular discovery workflows.
 White Paper
Life Science
White Paper
Life Science
Quick Reference Sheets
- Quick Reference Sheet
Force Field Builder
A one-page guide to calculate missing torsion parameters for ligands using the Force Field Builder panel.
- Quick Reference Sheet
Ligand Interaction Diagram
A one-page guide to using the Ligand Interaction Diagram for examining ligand-receptor interactions.
- Quick Reference Sheet
GlideMap
A one-page guide to using the GlideMap GUI for ligand placement guided by experimental density.

Latest insights from Extrapolations blog

With FEP+, “The Experiment is the Limit.”
Over the past century, small molecule drugs have represented the dominant modality in drug research, enabling medical breakthroughs that have saved countless lives.

Tackling Drug Solubility: AbbVie and Schrödinger Collaborate to Advance Accurate Prediction Methods
The complexity and size of drug candidates has grown in recent years as scientists pursue novel targets once considered undruggable.
 Blog
Blog
Can AlphaFold Models be Used for Structure-Based Drug Design? A Perspective Two Years In
We recently sat down with Edward Miller, Senior Director of Protein Structure Modeling at Schrödinger, to discuss his experience using AlphaFold models for SBDD.
Training & Resources
Online certification courses
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Free learning resources
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Other Resources