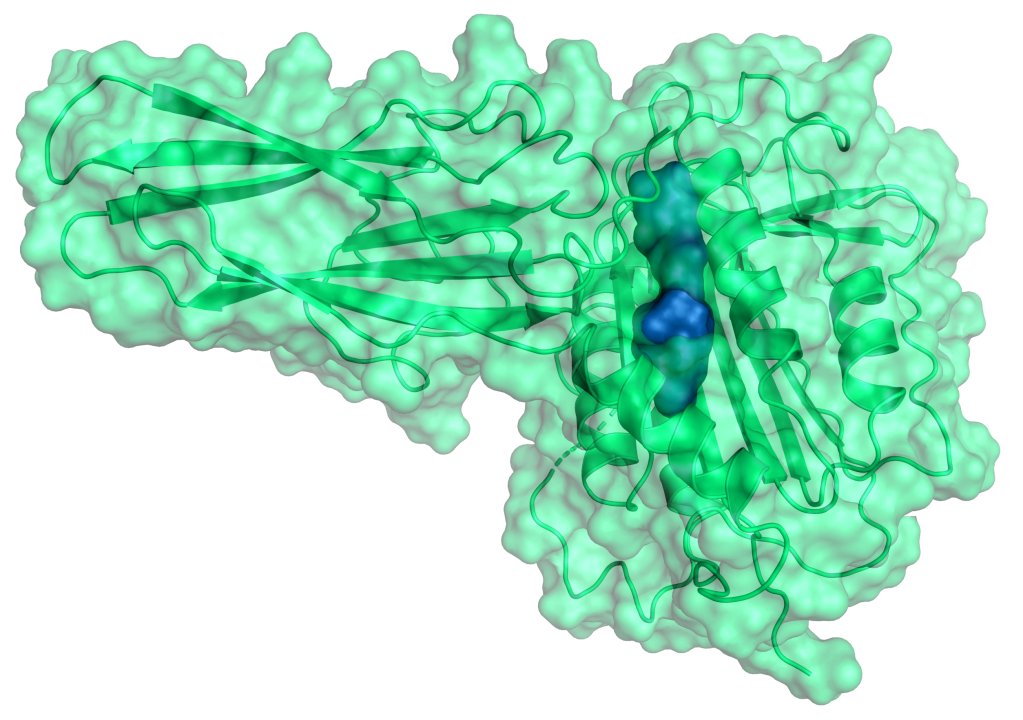
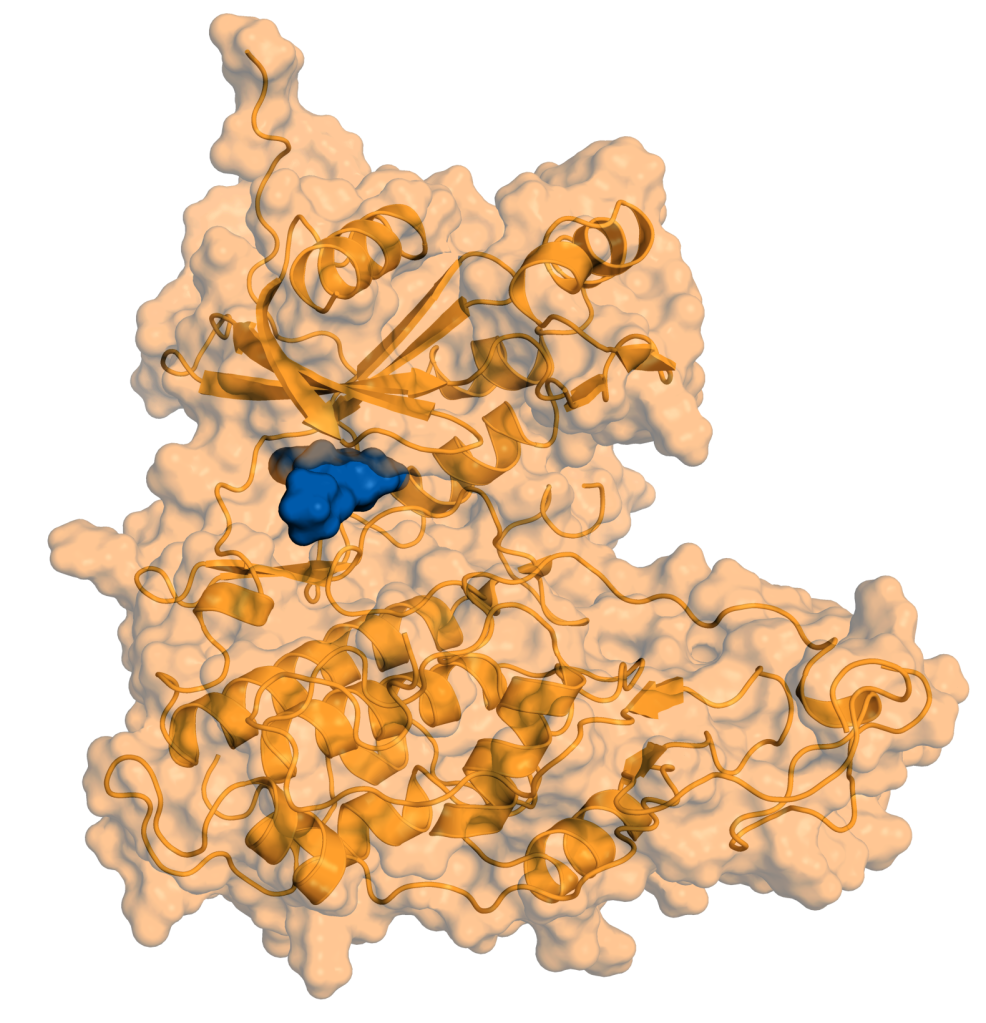
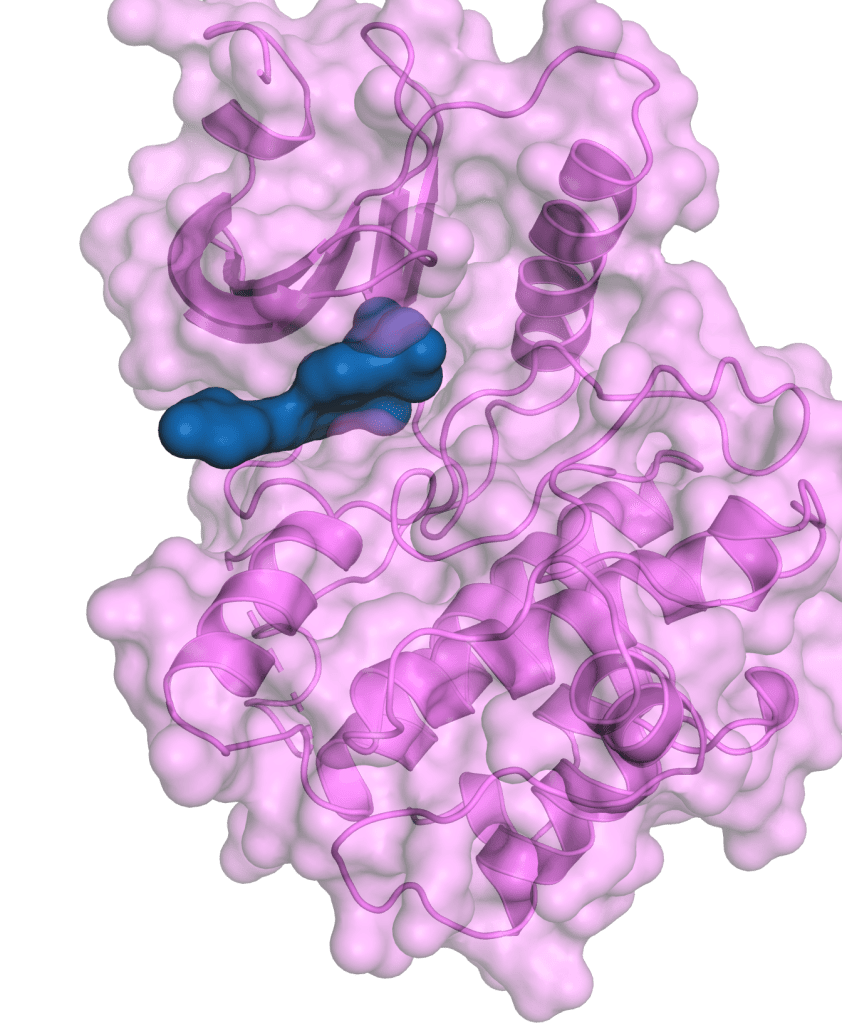
Therapeutic Pipeline
For more than 30 years, we have invested in advancing the science underlying our computational platform. As our technology advanced, our company evolved along with it.
Recognizing the opportunity to leverage the full power of our platform to drive internal discovery of molecules and clinical candidates for unmet medical needs, we formed Schrödinger’s therapeutics group.