Accelerating catalysis and reactivity r&d with atomic-scale simulation, machine learning, and enterprise informatics
As market demands evolve, R&D scientists across industries — from consumer packaged goods to automotives — face similar challenges in developing and optimizing the next-generation of catalysts. Scientists need new catalysts that help them reduce energy requirements, eliminate unwanted side products, and improve the selectivity and reactivity of reactions.
Solution Overview
We need new materials, and we need them fast. Fortunately, the continual march of scientific progress promises faster answers than in the past. Computer-driven molecular design, with its ability to generate massive quantities of simulated data, facilitates entry into new frontiers of chemical discovery for sustainable materials design. It brings the promise of speed and accuracy, and it allows R&D scientists to scan through large molecular space to triage down and experimentally test only the most promising chemistries.
Schrödinger’s Materials Science platform leverages the power of physics-based simulation, machine learning, and enterprise informatics to enable the optimization and discovery of effective and selective catalysts and reactive systems by offering:
- Differentiated model builders
- Interactive visualization and analysis tools
- Highly-efficient density functional theory (DFT) engines
- Customizable automated reaction workflows
- A collaborative enterprise platform solution
Application Overview
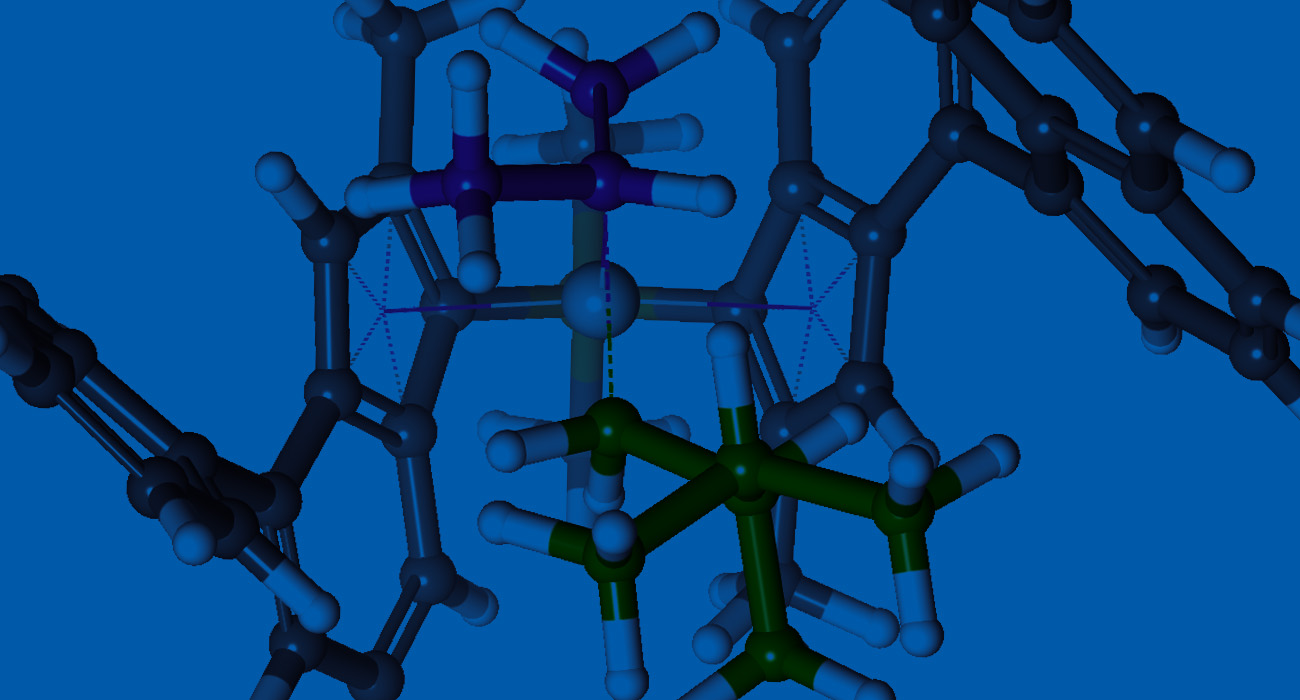
Catalysts
- Advance the knowledge of catalytic mechanisms: Simulate catalytic pathways to fundamentally understand the forces providing reactivity and selectivity
- Improve the lifetime of catalysts: Predict stability and degradation of catalysts and products
- Design and optimize catalysts: Automatically run high-throughput screening of catalysts by leveraging known mechanistic pathways with novel catalysts
- Benefit from inherently chemically agnostic workflows: Accelerate and automate any reaction mechanism in both homogeneous and heterogeneous screening workflows
Reactive Systems
- Understand reaction mechanism and pathways: Simulate reaction thermodynamics, kinetics, reaction rates, and barriers
- Accelerate product development: Automatically predict selectivity and activity of reactions with a library of interesting substrates and catalysts
- Optimize product properties: Compute tacticity of polymers with relative reaction pathway energies
- Predict product distribution: Automatically locate transition state
- Drive innovation in reaction design: Study effect of steric, electronic, and ligand on the reactivity
Team Collaboration and Digital Data Management
- Empower team collaboration: Employ webbased enterprise informatics tools for sharing experimental and predictive models seamlessly
- Amplify research with improved decisionmaking: Rapidly deploy automated reaction workflows and machine learning models to drive large-scale predictions and assist novel design approaches
- Improve project management: Accelerate project communication and collective learning by capturing, analyzing, and testing new ideas and data in a centralized platform
Products
Discover the Schrödinger products that enable your success in the catalysis industry.
Selected publications
-
Iron-catalysed Synthesis and Chemical Recycling of Telechelic 1,3-enchained Oligocyclobutanes.
Chirik P. J. et al. Nature Chemistry 2021, 13, 1 56-162.
-
Exploring the Mechanism of Cr(VI) Catalyzed Hypochlorous Acid Decomposition.
Busch M et al. ChemCatChem 2022, e202101850.
-
Intramolecular Hydroxyl Nucleophilic Attack Pathway by a Polymeric Water Oxidation Catalyst with Single Cobalt Sites.
Sun L. et al. Nature Catalysis 2022, 5, 414–429.
-
Olefin Metathesis Catalyzed by a Hoveyda– Grubbs-like Complex Chelated to Bis(2- mercaptoimidazolyl) Methane: A Predictive DFT Study.
Martínez J. P. et al. J. Phys. Chem. A 2022, 126, 5, 720–732.
-
Mechanistic Study of Metal–Ligand Cooperativity in Mn(II)-Catalyzed Hydroborations: Hemilabile SNS Ligand Enables Metal Hydride-Free Reaction Pathway.
Baker R. T. et al. ACS Catal. 2021, 11, 15, 9043–9051.
-
Cut-off Scale and Complex Formation in Density Functional Theory Computations of Epoxy-Amine Reactivity.
Laurikainen P. V. et al. ACS Omega 2021, 6, 44, 29424–29431.
-
Decarbonylative Fluoroalkylation at Palladium(II): From Fundamental Organometallic Studies to Catalysis.
Sanford M. S. et al. J. Am. Chem. Soc. 2021, 143, 44, 18617–18625.
-
One-Pot Chemo-bioprocess of PET Depolymerization and Recycling Enabled by a Biocompatible Catalyst, Betaine.
Kim K. H. et al. ACS Catal. 2021, 11, 7, 3996–4008.
-
Automated Transition State Search and Its Application to Diverse Types of Organic Reactions.
Friesner R. A. et al. J. Chem.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.