Battery and energy storage materials
Background
The design and manufacturing of safer, less expensive, and more effective energy storage devices is a critical challenge in a wide variety of industries including the automotive, aviation, and energy sectors with societal and environmental implications. Atomic-scale materials modeling has become an essential tool for the development of novel battery components — cathodes, anodes, and electrolytes — that support higher power density, capacity, rate capability, faster charging, and improved degradation resilience. Schrödinger’s Materials Science software platform provides a powerful atomic-scale modeling solution for comprehensive analysis of ion diffusion, mechanical response, and electrochemical response in electrodes and electrolytes, dielectric properties of potential electrolyte compounds, and other relevant properties.
Application: Virtual screening for sei forming li-ion battery additives
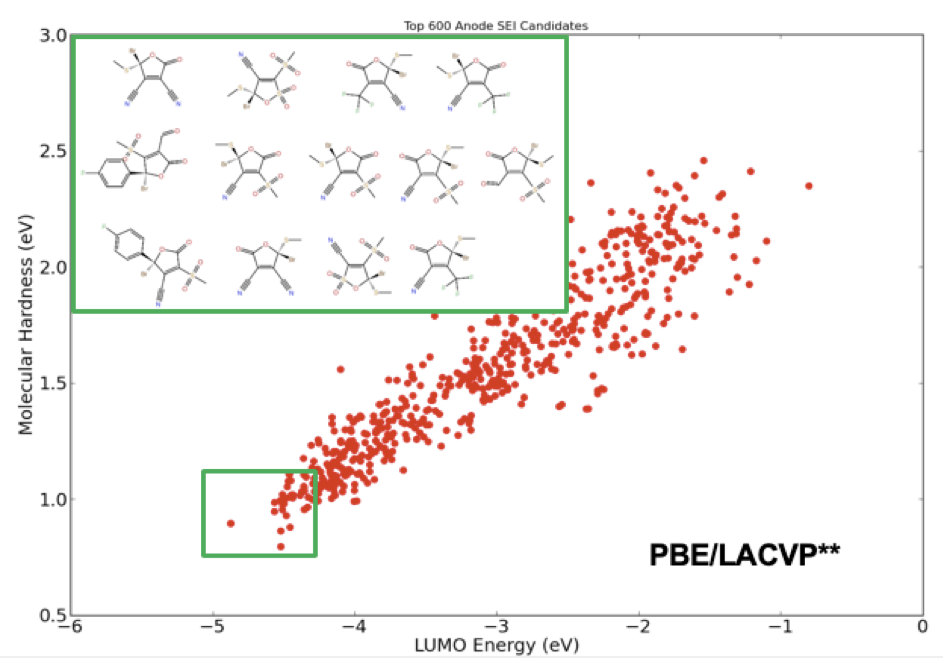
Additives in electrolyte formulations are designed to increase dielectric strength and electrode stability by facilitating the formation of a stable solid electrolyte interphase (SEI). To enhance stability of the SEI, these electrolyte additives should exhibit high reduction potential and increased reactivity. From a molecular design perspective, the former is related to the energy of the lowest-unoccupied molecular-orbital (LUMO), while the latter is correlated with molecular hardness. Using these two key molecular descriptors, one can screen through a large library of compounds to find the ideal candidates for new electrolyte additives. Figure 1 illustrates the final stage of a virtual screening example applied to an automatically generated library of 368,073 unique electrolyte compounds with 6 cyclic carbonate and sulfone cores and 18 R-groups. The final list of compounds showcases a chemical design space pregressively screened using the accurate force field (OPLS2005 [1]), and semiempirical quantum chemistry (RM1 [2]), followed by a final selection process based on a full quantum chemical analysis with density functional theory (DFT).
Application: Dielectric properties of molecular electrolytes
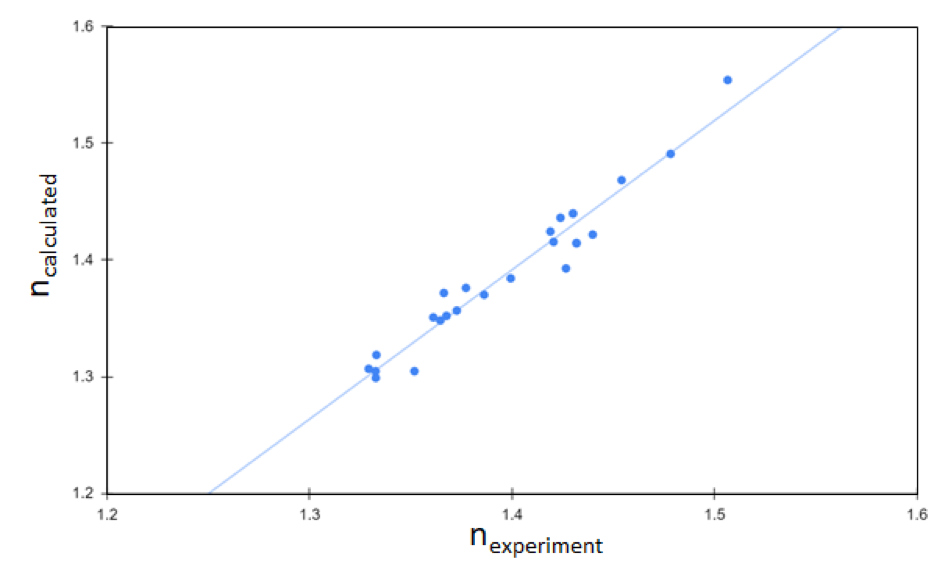
The dielectric constant is another key design factor for battery electrolytes. Since the dielectric constant correlates directly with optical response for compounds with relatively low polarity, screening for compounds with high refractive index using the Schrödinger Materials Science Suite can quickly narrow down the list of potential molecular electrolytes to those with desirable dielectric properties. A study summarized in Fig. 2 showcases the accuracy of computational prediction for refractive indices of molecular liquids. As shown in Fig. 2, the calculated dielectric constants agree well with experimentally measured values [5], validating the accuracy of theoretical predictions. This predictive capability is a direct result of (1) the speed and accuracy of compute engines used by Schrödinger software and (2) the seamless integration between two independent simulation methods – quantum mechanics (QM) and classical molecular dynamics (MD). This example shows how theoretical predictions at the atomic-scale assess key intrinsic properties of battery materials, such as dielectric properties.
Application: Intercalation energetics of electrode materials
Ion intercalation in electrodes is a critical process that determines the core performance of rechargeable batteries. The Schrödinger Materials Science platform’s periodic DFT engine, Quantum ESPRESSO, has proven its scientific value over a wide range of applications, including the analysis of thermodynamics and kinetics of ion intercalation processes in electrodes.
Work by Arroyo-de Dompablo and coworkers [3] apply Quantum ESPRESSO to design and characterize LiMXO4 cathode materials based on computationally predicted intercalation voltages [3]. Schrödinger’s comprehensive list of modeling and simulation workflow solutions coupled with Quantum ESPRESSO helps to extend these types of analysis to cover broad chemical space and properties to facilitate the research of novel electrode materials.
Apart from intercalation potentials, there is a number of other electrode properties that can be studied with computational methods. Some important examples include ion migration kinetics and pathways, mechanical properties, the volume change upon ion intercalation, electronic properties, chemical stability, and reactivity with respect to electrolyte.
Application: Analysis of polymer electrolyte using classical molecular dynamics
Faster diffusion of lithium ions in battery electrolytes is desirable to improve battery performance. In a recent publication [4], Brooks and coworkers performed thorough investigations of lithium ion diffusion in polyethylene oxide (PEO) electrolyte. Using the Schrödinger Materials Science platform and its classical MD workflows, the authors studied the key diffusion mechanisms of lithium ions in a PEO electrolyte mixed with bistriflimide (also known as TFSI) anions. The lithium ion diffusion coefficient was examined with varying temperature and Li:TFSI ratio [4]. Analysis of the mean square displacement diffusion rates and Li/polymer coordination suggest that PEO is too flexible causing it to bind the lithium ions too tightly, which implies that a less flexible polymer blend used for electrolyte could lead to a faster lithium ion diffusion.
Apart from the ion diffusion itself, the details of coordination between the ion and the electrolyte can be captured and compared, helping to guide research to identify the best chemical groups. Thermophysical and mechanical properties of the polymer electrolytes are also accessible through molecular dynamics simulations expanding the performance criteria of solid electrolytes that can be considered in early screening.
Summary
Atomic-scale simulation and modeling technologies integrated within Schrödinger’s Materials Science software provide critical insight in all facets of the materials design process for battery components- electrolytes, electrodes, and formation of stable SEI. Schrödinger’s comprehensive list of solutions can elucidate key chemical processes of the materials and characterize their crucial thermophysical properties, which can boost the cost effectiveness of the design pipeline for novel materials and accelerate the development of novel energy storage devices.
Selected publications
-
Banks, Jay L., et al. “Integrated modeling program, applied chemical theory (IMPACT).”
Journal of computational chemistry 26.16 (2005): 1752-1780.
-
Rocha, Gerd B., et al. “RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I.”
Journal of computational chemistry 27.10 (2006): 1101-1111.
-
Arroyo-de Dompablo, M. E., et al. “On-demand design of polyoxianionic cathode materials based on electronegativity correlations: An exploration of the Li2MSiO4 system (M= Fe, Mn, Co, Ni).”
Electrochemistry Communications 8.8 (2006): 1292-1298.
-
Brooks, Daniel J., et al. “Atomistic Description of Ionic Diffusion in PEO–LiTFSI: Effect of Temperature, Molecular Weight, and Ionic Concentration.”
Macromolecules 51.21 (2018): 8987-8995.
-
CRC Handbook (85th Ed); Kim, Sunghwan et al. “PubChem 2019 update: improved access to chemical data.” Nucleic acids research vol. 47,D1 (2019): D1102-D1109. ; Gregory, Andrew, R. N. Clarke.
“Tables of the complex permittivity of dielectric reference liquids at frequencies up to 5 GHz.” National Physics Laboratory MAT 23 (2011).
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.