
Free energy calculations are revolutionizing how early-stage drug-discovery campaigns are undertaken. In this short, 10-minute video, Schrödinger Senior Principal Scientist, Dr. Jennifer Knight, demonstrates the ability of Protein Residue Mutation FEP+ to elucidate ligand binding preferences for large families of off-targets and how, on an active drug discovery project, this strategy rapidly rescued a series by dialing out off-target liabilities.
Stories from drug discovery: Dialing out off-target liabilities with protein residue mutation FEP+ in an active drug discovery project

Summary
Jennifer Knight is a Senior Principal Scientist in Schrödinger’s Drug Discovery Group. While working on one of their active drug discovery projects, the team came across some unexpected challenges with selectivity.
The Drug Discovery Group was working on a project where a competitor series for the same target had demonstrated potential liabilities due to several off-target kinases. The team identified their own series and used structure-based modeling with ligand FEP+ to optimize potency and improve selectivity over the known off-targets.
Over the course of seven months, the team profiled over six million design ideas. We triaged the designs using a variety of chemistry filters, Glide docking, and Active Learning strategies. Ten thousand went through the full ligand FEP+ calculations and multiple series and subseries were identified that looked promising. However, these new series picked up many other unanticipated and problematic off-target liabilities as well.
Instead of abandoning the project (and having months of research go to waste), the team decided to explore further. Looking into the binding pocket, the team observed a particular pattern—compounds in one series tended to hit kinases that had a particular amino acid at a given position, while compounds in another series tended to hit kinases that had a different amino acid at that position.
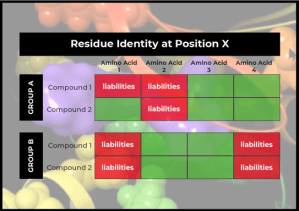
This pattern gave the team an idea: What if they could use Protein Residue Mutation FEP+ as a way of accounting for these overall kinome scan profiles? Could they use the information (the amino acid identity at that one position) to understand how the compound might interact with all kinases that have that particular amino acid at that position?
The team found that, indeed, Protein Residue Mutation FEP+ was able to recapitulate the trends observed in the experimental data. This strategy allowed the team to reliably identify which family of kinases would be problems for their compounds and which ones wouldn’t. So, the team used this modeling approach prospectively to profile new design ideas and identified compounds that were predicted to not hit any other of these kinase families.
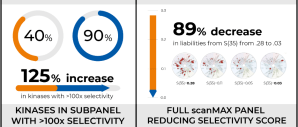
In a single round of synthesis, the team was able to drastically reduce the number of kinases that were hit by their series, and substantially minimize the off-target liabilities. This approach resulted in dramatically improved selectivity as observed in a preliminary panel of 20 kinases (increasing the percentage of kinases from 40% to 90% with >100x selectivity window) and in the full scanMAX panel (reducing the selectivity score, S(35), from 0.28 to 0.03). Both on-target ligand FEP+ and protein FEP+ for off-target selectivity were used successfully in this novel strategy to rescue these project series.
By using the combination of these two methods—determining not only the compounds with the best on-target potency with Ligand FEP+, but also eliminating the compounds that had off-target liabilities by using Protein FEP+—the team identified the best compounds to move forward into synthesis in approximately three months.