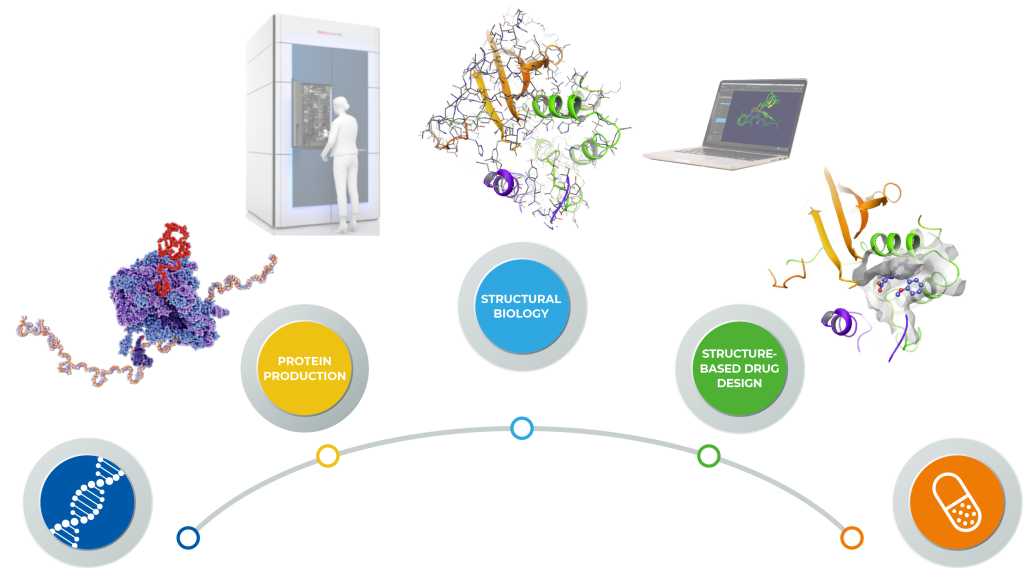
An expedited gene-to-drug approach using thermo scientific cryo-em and the Schrödinger platform
Introduction
Structure-Based Drug Design (SBDD) imparts cost-efficiency, timeliness and superior properties to target-based small molecule drug discovery1-3. Today, computational approaches have evolved to the point where they provide quantitative predictions of ligand affinity4 and selectivity5. Historically, X-ray crystallography has driven SBDD efforts resulting in the development of potent and selective protease and kinase inhibitors for the treatment of a variety of diseases including AIDS6 and cancer2. However, many pharmaceutically important drug targets such as large macromolecular assemblies or membrane proteins are less amenable to SBDD due to the lack of 3D structural information. Recent advances in protein production and cryo-electron microscopy (cryo-EM) have expanded the role of SBDD for these critical targets7,8. Here, we present the approach taken by Thermo Fisher Scientific and Schrödinger research teams that deployed GeneArt Gene-to-Protein, Thermo Scientific iSPA Workflow and the Schrödinger Drug Discovery platform.
Target Selection
Selection of targets for initiation of drug discovery programs is dependent on an understanding of the unmet medical need and multiple lines of evidence from clinical, genetic and preclinical studies that inform therapeutic potential (Figure 1). Biological rationale in the form of biochemical, pharmacological and genetic data that link modulation of target activity to pathophysiology and disease mechanisms are key factors. For example, targets with a clearly defined relationship between genotype and phenotype in preclinical models and human studies are considered higher priority. Confirming the impact of inhibition or activation and an understanding of tractability are also important aspects of target validation that serve as triggers for program initiation.
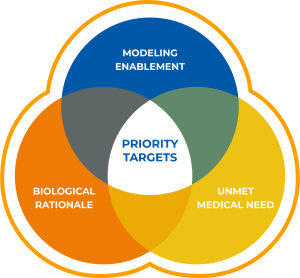
All targets must be modeling enabled for SBDD. After a careful review of current clinical and preclinical programs for a given target, challenges that could be solved by Schrödinger’s computational platform are identified. The Schrödinger platform is used to analyze protein structure quality and binding site druggability, followed by an assessment of the amenability of the structures for use with the technology. Due to the lack of high-quality structures, a large number of very interesting targets that meet the criteria for biological rationale and therapeutic momentum would need to be reprioritized until high-resolution structures are available. This is often because such targets are membrane proteins or proteins which form large multimeric structures that are often challenging for X-ray crystallography. Fortunately, these types of targets are often well suited for structural determination by cryo-EM, and allow for modelling of previously unreachable targets. A target was selected for the collaboration after careful analysis of the biological rationale, the unmet medical need and the potential for cryo-EM enablement of the target.
Protein Production
Once a target has been selected, the first hurdle to overcome is the production of protein of suitable quality for use with cryo-EM. Cryo-EM is typically applied to proteins that are multimeric and/or membrane protein complexes with a molecular weight of over 50 kDa. This poses challenges for protein production and purification, particularly within the timeline expectations of drug discovery. Thermo Fisher’s GeneArt platform comprises a Gene-to-Protein service that only requires a protein sequence and covers every step from gene synthesis to protein purification (Figure 2).

In this project, the process involved gene optimization, DNA synthesis, and transient protein expression in Gibco Expi293 cells. Purification from the cytoplasmic fraction via a terminal His tag yielded protein that was highly pure and required no further purification. The entire workflow from gene sequence to purified protein was completed within 6 weeks. The protein was obtained at a concentration of 5mg/ml, sufficient for creating dense micrographs with many particles for subsequent analysis, and with a yield in excess of 10 mg, more than enough protein to supply a cryo-EM based structure determination pipeline for months.
Cryo-EM Structure Determination
For structure determination of our selected target, we used the Thermo Scientific iSPA Workflow, a commercially available single particle analysis (SPA) solution for drug discovery. It includes a Thermo Scientific Vitrobot Mark IV device for the preparation of vitrified cryo-EM specimens, which facilitates rapid plunge-freezing of holey carbon grids in liquid ethane after application and blotting of the protein solution. This achieves embedding of proteins in a thin layer of non-crystalline ice, preserving their native state in solution. The specimens are then subjected to cryo-EM data collection performed on a Thermo Scientific Krios Rx cryo-TEM. The Krios Rx is operated with Thermo Scientific EPU, which is data acquisition software that enables automated screening and data collection across multiple grids (thanks to the recent EPU Multigrid feature). The Krios Rx records movies that represent 2D projection images of the target of interest and are subsequently used for computational 3D image reconstruction. After data collection, 3D reconstruction involves orienting and averaging hundreds of thousands of 2D images of isolated particles to calculate a high-resolution map of the protein. EPU Quality Monitor and EPU Data Management (powered by Thermo Scientific Athena Software) ensure optimal data quality and data flow.
In this study, the iSPA Workflow readily yielded ~2.5 Å resolution reconstructions for both unliganded and liganded complexes (Figure 3). The initial structural enablement of this target required two weeks from receipt of protein, involving two imaging attempts. The first attempt, where protein was frozen at a concentration of 0.5 mg/ml, was not successful. Cryo-EM screening yielded an uneven distribution of particles, which were only found on carbon areas or close to the edge of the carbon film holes. Since high particle density is generally favorable for vitrification, GeneArt delivered a second batch of protein at a higher concentration (5 mg/ml) which resulted in an even and highly dense “monolayer” distribution of particles on the grid. Compounds of interest were dissolved in DMSO and added to the protein solution prior to vitrification, aiming for final compound and DMSO concentrations of ~50 μM and ~0.5%, respectively. For each dataset, we collected roughly five thousand movies and Relion 39, running on a low-cost quad-GPU workstation, was used for the reconstruction. In total, the time from sample preparation to high-resolution reconstruction of the first complex structure was performed within 3 days with the ability to solve additional liganded structures on a similar timeframe.
 |
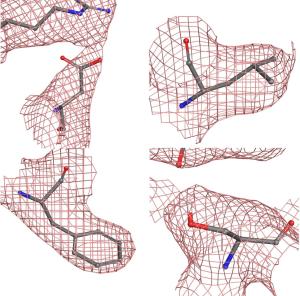 |
Figure 3. Left, the Thermo Scientific Krios Rx Cryo-TEM high-end microscope is the first pharma-dedicated solution for cryo-EM SBDD with a guaranteed high throughput ideal for iterative structure determination. Right, detail of 4 representative residues showing the coulombic potential reconstructed from the EM data collection and the fitted model. As can be seen, the data was sufficient to ascertain the position of most sidechains.
Structure-Based Drug Discovery
Once protein-ligand complex structures were obtained, atomic models were prepared with the Schrödinger platform and passed into the Schrödinger SBDD pipeline. As part of this process, ligands were placed using GlideEM, a tool that combines molecular-mechanics-based docking with real-space cross correlations to place ligands into cryo-EM maps10. The resulting atomic models were then refined with Phenix/OPLS3e (the prior version of OPLS4) which combines state-of-the-art real-space refinement with advanced Schrödinger force fields and implicit solvent models that can capture the underlying physics of the protein-ligand system. This workflow is able to rapidly create robust atomic models that are consistent both with the cryo-EM data and the physics of the system.
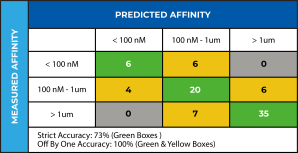
These atomic models can then be leveraged within the Schrödinger SBDD pipeline to virtually screen massive numbers of diverse compounds. In addition, targeted computational screens can be designed to remove known liabilities or exploit opportunities to differentiate a series from competitor compounds. In this case, we used the refined atomic model with FEP+ to create an affinity prediction model that was validated by retrospectively predicting affinity differences for a previously patented 62-compound congeneric series. We were able to capture several major affinity cliffs, thus validating that the refined atomic models were of sufficiently high quality to be used in prospective design as part of an SBDD-led program (Figure 4). We used this validated affinity model to explore modifications to the compounds to address the target product profile goals identified by the project team, such as novelty, improving permeability, and elimination of a potentially reactive group, all while maintaining potency. Pathfinder was used to ideate promising compounds, Glide was used to generate potential poses and FEP+ with our validated affinity model was used to score potency. Within weeks of obtaining the cryo-EM structure of the target-ligand complex, structure-based computational methods were used to prioritize compounds for synthesis.
Conclusion
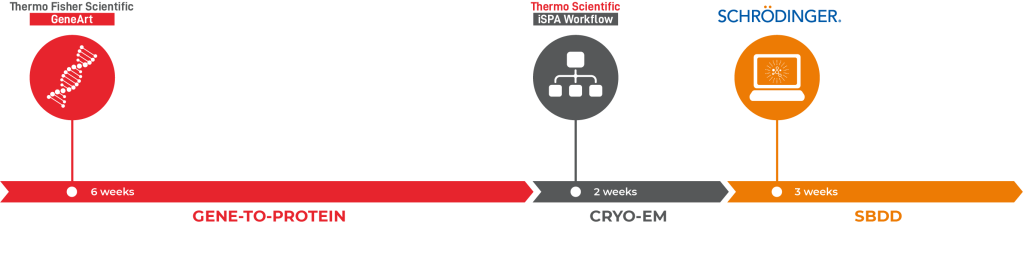
Using a combination of solutions from Thermo Fisher’s GeneArt Gene-to-Protein, Thermo Scientific iSPA Workflow (Thermo Scientific Cryo-EM) and the Schrödinger Drug Discovery platform, the team was able to facilitate the structural enablement of the drug target within two months and arrive at novel, computationally-designed chemical matter just a few weeks later (Figure 5). This clearly illustrates that the combination of cryo-EM and computational chemistry methods can create a pipeline that can have a major impact on drug discovery projects.
References
-
RCSB Protein Data Bank: Enabling Biomedical Research and Drug Discovery
Goodsell DS et al. Protein Sci. 2020, 29, 52–65
-
Structural Biology Contributions to Tyrosine Kinase Drug Discovery
Cowan-Jacob SW et al. Curr Opin Cell Biol. 2009, 21, 280-287
-
Structural Biology Contributions to the Discovery of Drugs to Treat Chronic Myelogenous Leukaemia
Cowan-Jacob SW et al. Acta Crystallogr. 2007, D63, 80-93
-
Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects
Schindler CEM et al. J Chem Inf Model. 2020, 60, 5457-5474
-
Is Structure-Based Drug Design Ready for Selectivity Optimization?
Albanese SK et al. J Chem Inf Model. 2020, 60, 6211-6227
-
Molecular Basis for Drug Resistance in HIV-1 Protease
Ali A et al. Viruses. 2010, 2, 2509-2535
-
The Rapidly Evolving Role of Cryo-EM in Drug Design
Wigge C et al. Drug Discovery Today: Technologies in press, 2020, 38, 91-102
-
Multiparameter RNA and Codon Optimization: A Standardized Tool to Assess and Enhance Autologous Mammalian Gene Expression
Fath S et al. PLoS ONE, 2011, 6, e17596
-
New Tools for Automated High-resolution Cryo-EM Structure Determination in Relion-3
Zivanov J et al. eLife, 2018, 7, e42166
-
GemSpot: A Pipeline for Robust Modeling of Ligands Into Cryo-EM Maps
Robertson MJ et al. Structure, 2020, 28, 707-716