How machine learning enables accurate prediction of precursor volatility

Challenges in predicting volatility
A crucial process in manufacturing CPUs and other high-tech devices is the deposition of solid material from reactive vapors. Different precursor vapors are used for chemical vapor deposition, vapor phase epitaxy, atomic layer deposition – and indeed the reverse process of atomic layer etching – with the precursor chemistry carefully designed for each case so as to control material quality at the nanoscale. But what all these techniques have in common is that the precursor chemicals must evaporate or sublime at a low enough temperature. Too much heating when vaporizing a precursor can make it decompose, causing it to be undeliverable to the growing surface.
With volatility playing such a central role in this technology (and in other fields like distillation, refrigeration, inkjet printing, food and perfumes), it is surprising that we understand so little about it. Volatility is the product of a remarkably fine balance of interatomic forces, dictating the extent to which molecules condense together as a solid or liquid, or bounce apart into a vapor and deliver a certain vapor pressure at any given temperature. These interatomic forces can be computed very precisely with quantum mechanics for one molecule or a group of molecules, but not at the scale of a liquid or solid. Even with today’s computing power, routinely and accurately predicting precursor volatility ‘from first principles’ remains unfortunately out of reach.
Machine learning approaches
Could an alternative more empirical approach prove useful? Does enough experimental data exist to find the relation between volatility and chemical structure? The vaporization of some organic molecules, such as alcoholic fractions or natural fragrances, has been of interest for centuries and high-quality vapor pressure data are available in the literature. Over the last decade, these data have been analyzed with advanced fitting algorithms that come under the umbrella of ‘machine learning’. Schrödinger has leveraged the latest machine learning techniques to develop a highly-accurate model that predicts the volatility of organic molecules up to C20.
However, when building machine learning models to predict volatility of precursor molecules, which are typically organometallic complexes, the situation is not so straightforward. New precursor molecules are constantly being proposed and evaluated. Commercial sensitivity sometimes means that data are partially withheld, or plagued by experimental configuration differences from laboratory to laboratory. Additionally, for the common aim of material processing, complete pressure-temperature curves are rarely measured, as it is more pragmatic to focus on the temperature for vapor to transport successfully to the reactor. As a result, datasets for building predictive models are sparse and incomplete.
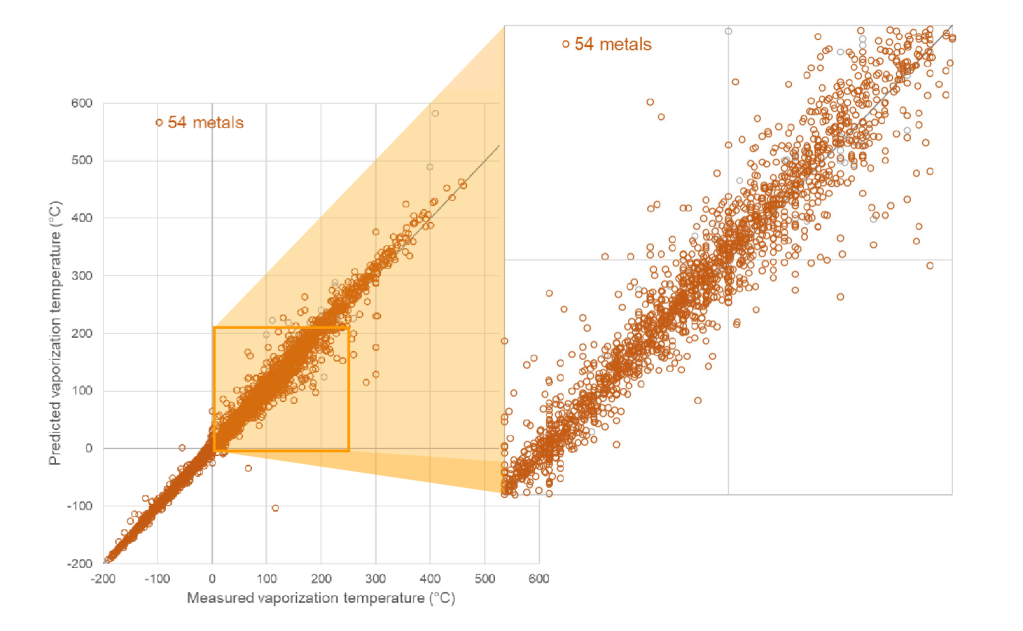
Prediction of volatility for inorganic and organometallic complexes
Schrödinger scientists embarked on the challenge of building machine learning models to predict the volatility of precursor molecules. Using in-house expertise in machine learning and advanced informatics, Schrödinger scientists collated and digitized information about organometallic precursors from disparate literature sources and applied a variety of machine learning algorithms (such as Random Forest and Neural Networks) in conjunction with different chemoinformatic descriptors and fingerprints. The result is the first capability of its kind for accurately and efficiently predicting the volatility for inorganic and organometallic complexes from their chemical structures. For complexes of the fifty most common metals and semimetals, the model predicts the evaporation or sublimation temperature at a given vapor pressure with an average accuracy of ±9°C (which is about 3% of the absolute temperature). As a trained model, the turnaround time is fast with the ability to compute hundreds of complexes per second.
New avenues for precursor development
This predictive model opens a new path for designing novel precursors with improved performance, not only improving their deposition or etch chemistry, but also optimizing the temperature at which they evaporate or sublime and can be delivered as a vapor. This advance will allow a much wider range of structural modifications to be screened computationally than before and will produce candidate precursors for experimental synthesis and testing that are both less risky and more innovative. This volatility model, together with Schrödinger’s quantum mechanics-based workflows for computation of reactivity and decomposition, gives scientists a complete design kit for vapor-phase deposition or etch, delivering a faster pace of research into materials and processes for new technologies.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.