Innovation in atomic-level processing with atomistic simulation and machine learning
Background
The Schrödinger Materials Science Suite offers computational tools specifically tailored to studying the gas-surface chemistries of atomic layer deposition and related nanofabrication processes. Schrödinger software is designed for rapid and automated enumeration of chemical space, detailed study of gas-surface chemistries at the quantum mechanical level (usually with density functional theory, DFT) and prediction of key properties. This modeling approach thus has a unique role to play both in deepening our understanding of existing processes and in discovering novel chemicals.
Many of today’s high-tech devices are manufactured by processing materials at the nanoscale, including computer chips, data storage and communications devices, sensors, solar cells and batteries. Making devices smaller, more powerful and more energy-efficient means developing new patterning, deposition and etch techniques at ever-finer resolution, in some cases down to just a few atoms thick. Chemical processing is being pushed to its limits in terms of purity, uniformity, conformality and substrate-selectivity.
Optimizing a deposition or etch process to more stringent tolerances requires a deep understanding of the underlying chemistry. Integrating new materials into a device often requires the discovery of new chemicals with improved properties. In both respects, researchers are increasingly turning to computer simulations to cut down on lab work and make discoveries faster.
The Schrödinger Materials Science Platform offers computational tools specifically tailored to tackling this problem, by studying the gas-surface chemistries of deposition, etch and related nanofabrication processes. Schrödinger software is designed for rapid and automated enumeration of chemical space, detailed study of gas-surface chemistries at the quantum mechanical level (with density functional theory, DFT) and prediction of key properties. This modeling approach can both deepen our understanding of existing processes and help discover novel chemicals.
Automate Precursor Screening for Improved Properties
Organometallic complexes can be used to deposit or etch pure metals or compounds such as oxides or nitrides. However, synthesis of the complexes in the lab can be time-consuming and difficult because many complexes react violently in air. Subsequent characterization and testing in a reactor is even more costly. Digital chemistry can rapidly screen larger numbers of chemicals than can ever be experimentally tested, narrowing down the chemical space and discovering the most promising candidate molecules for deposition or etch.
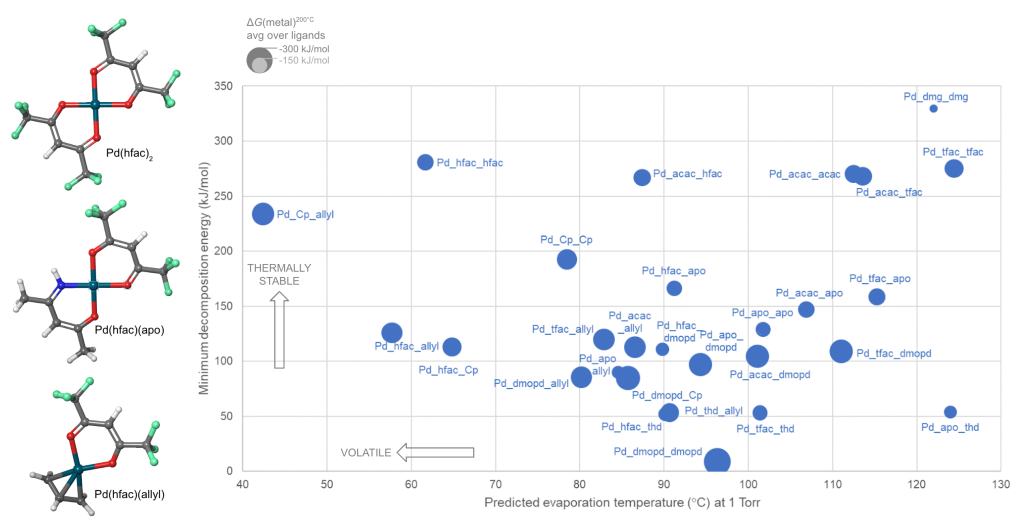
Schrödinger provides easy-to-use software for automated generation and assessment of hundreds or thousands of precursor gases. A library of hundreds of commonly-used ligands is provided, including bidentate and haptic ligands, and the library can be customized. A selection from the library is then used to enumerate and build all possible precursor complexes. We present here a case study in which a library of 10 ligands form 55 distinct complexes with divalent palladium (Pd) as the metal cation and some of these candidate complexes are shown in Figure 1.
Synthesizability
Combining two or more different ligands in a heteroleptic complex is a promising way to tune precursor properties, but it is difficult to guess whether a given combination can in fact be synthesized. This question can be answered by computing the energy for ligand exchange back to homoleptic complexes. In the Pd example, this reveals that 14 of the heteroleptic complexes are unstable and can not be synthesized.
Reactivity
Precursors for deposition must be carefully designed so as to react cleanly and deposit the desired solid material, via thermolysis or plasma activation (chemical vapor deposition, CVD) or via reactions with adsorbed fragments (atomic layer deposition, ALD). Likewise, reagents for etching must react with surface atoms to form gaseous products. Lists of candidate precursor molecules can be efficiently screened against deposition or etch chemistries using Jaguar, Schrödinger’s code for molecular DFT. Figure 1 displays the computed reactivity of the complexes with respect to Pd deposition.
Volatility and Thermal Stability
To be useful in a real CVD or ALD process, a precursor must also be volatile and thermally stable during storage and delivery. This limits the process to a window of viable temperatures, with insufficient vapor pressure at lower temperatures and impurities from decomposition at higher temperatures. For instance, Pd(thd)2 suffers from decomposition when heated to 200-250°C for the ALD of Pd metal.1, 2 Calculating volatility and thermal stability with Schrödinger’s tools gives information on how to tune these properties and widen the temperature window for maximum process flexibility.
Schrödinger has used machine learning (ML) to develop a unique model for predicting the evaporation or sublimation temperature of organometallic complexes at a given pressure. The ML-volatility model is accurate to an average of ±9°C over the training set and can evaluate hundreds of molecules per second.
The thermal stability of precursor gases can be quantified by considering the unimolecular decomposition channels: bond dissociation and β-H elimination. All such reactions in each molecule are automatically computed at the DFT level and the lowest energy reaction is selected as the most likely. The minimum decomposition energy of each complex is thus a measure of its thermal stability.
These approaches were applied to compute the properties of the Pd complexes, and Figure 1 shows how this allows the candidate precursors to be assessed.
Rational innovation is facilitated by Schrödinger’s atomistic modeling tools. Novelty is maximized by using a large library of ligands to build a wide variety of complexes, homoleptic and heteroleptic. Risk is minimized by selecting for lab study only those precursors computed to properly combine the desired properties of synthesizability, volatility, reactivity and stability.
Gain Insights Into Competing Processes for Optimum Deposition Conditions
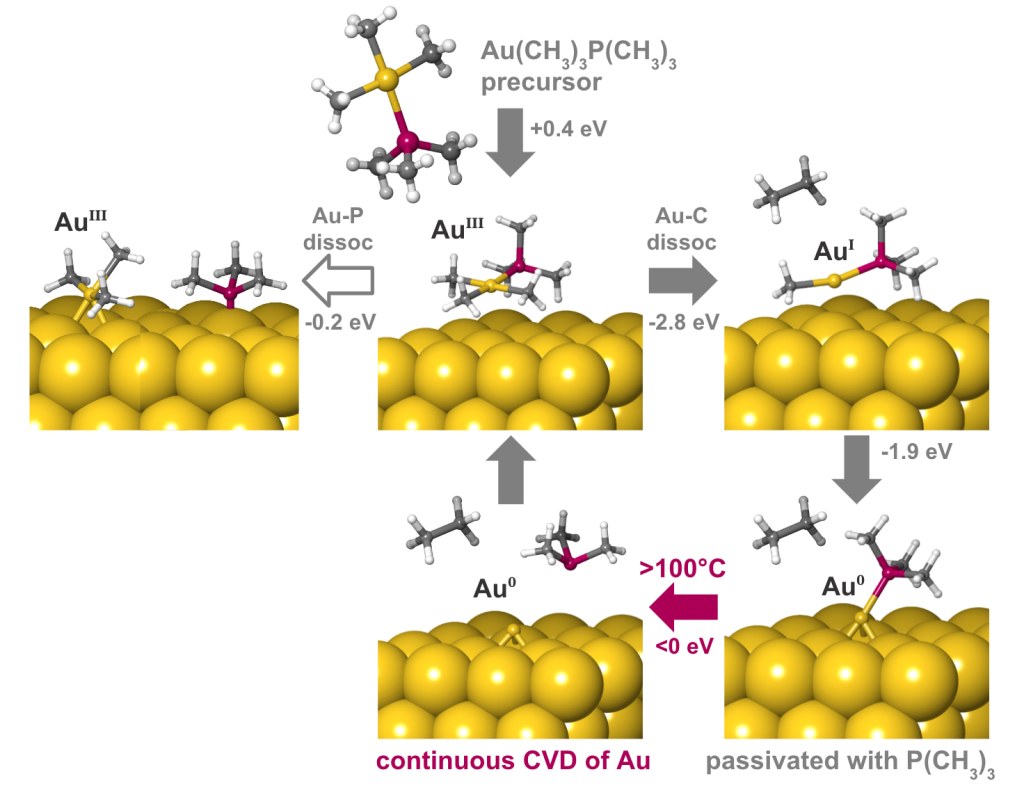
A deposition or etch process consists of a sequence of individual reaction steps, often in competition with other reactions. Simulations give atomic-level insight into these complicated mechanisms and how they depend on reactor conditions such as temperature and pressure. Here we investigate the mechanism of gold deposition from the single-source precursor trimethylphosphinotrimethylgold(III). Experiments show that gold films grow in a self-limiting fashion when this precursor is pulsed at low temperature, switching over to continuous CVD at 100°C and higher, but the reason is unknown.3
To explain this behavior, the Quantum ESPRESSO code for periodic DFT has been used to examine the detailed mechanism of reactive adsorption of the precursor onto a metallic gold surface. Thermodynamics of competing reaction steps have been computed as a function of process temperature and pressure with Schrödinger’s tools for adsorption and surface chemistry [4].
The results are summarized in Figure 2. The physisorbed precursor is in a metastable state, from which dissociation of Au-P or Au-C bonds can lead to chemisorption. Au-C dissociation and elimination of C2H6 is found to be the favored pathway, due to a strong thermodynamic driving force towards reduction of Au(III) to Au(I) and eventually Au(0). Trimethylphosphine is computed to bind to the surface, forming an adlayer that passivates against further precursor adsorption. This explains the low rate of self-limited deposition at <100°C. The computed free energies also reveal that desorption of trimethylphosphine becomes favored at higher temperatures, in agreement with the crossover to CVD seen experimentally at >100°C.
Finding out which bond breaks and how the surface is passivated opens up chemically-informed strategies for improving the process, for example by choosing a suitable co-reagent or modifying the precursor structure. Digital chemistry allows rational design to take the place of trial-and-error.
This example illustrates how changes in process behavior can be traced back to competing chemical pathways. DFT simulations give unique qualitative insight into atomic structure, bonding and oxidation states of the intermediates along each reaction pathway. In addition, quantitative information about kinetics and thermodynamics as a function of process temperature can be computed, giving valuable guidance on how to optimize the nanofabrication process. The result is improved control of purity, uniformity, phase and substrate-selectivity.
Conclusion
These cases illustrate how Schrödinger’s Materials Science Platform provides powerful and customizable computational solutions to accurately model surface reactivity, predict precursor properties and discover novel chemicals for optimized materials deposition or etch processes.
References
-
Low temperature atomic layer deposition of noble metals using ozone and molecular hydrogen as reactants,
J. Hämäläinen et al., Thin Solid Films 2013, 531, 243-250, DOI 10.1016/j.tsf.2013.01.091.
-
Atomic layer deposition of noble metals: Exploration of the low limit of the deposition temperature,
T. Aaltonen et al., J. Mater. Res. 2004, 19, 3353–3358, DOI 10.1557/JMR.2004.0426.
-
Reaction mechanism of the Me3AuPMe3–H2 plasma-enhanced ALD process, M. Van Daele et al.,
Phys. Chem. Chem. Phys. 2020, 22, 11903-11914, DOI 10.1039/C9CP06855D.
-
First-principles investigation of crossover between ALD and CVD in the thin film deposition of gold,
C. N. Brock. D. J. Giesen, A. Fonari & S. D. Elliott, Materials Research Society Spring Meeting, 2022.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.