Machine-learned force fields for improved materials modeling
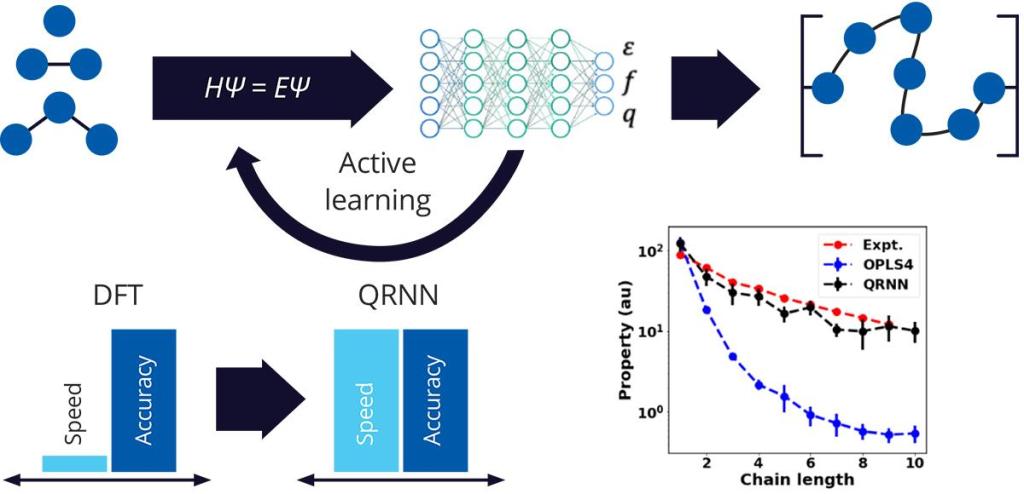
Machine-learned force fields (MLFFs) are designed to improve upon traditional force fields by incorporating machine learning models to accurately model the interactions between atoms and molecules. This technique is based on a neural network potential energy surface (NN-PES) architecture, where the model is trained to reproduce the total electronic energy of the system to chemical accuracy.
The NN-PES architecture is computationally efficient and utilizes local atomic descriptors to account for the surrounding environment. The model is trained by minimizing prediction errors against electronic energy obtained from any theory, such as density functional theory (DFT). This method enables simulations on larger length and time scales when compared to ab initio molecular dynamics (AIMD) methods, but with similar accuracy.
Previously, MLFF methods were limited to short-range interactions, but recent efforts have incorporated complex neural network architecture, charge recursive neural network (QRNN), that includes training of atomic charges to capture long-range interactions1. As a result, complex systems such as battery electrolytes can now be more accurately modeled with MLFFs.
Our combination of OPLS4 for initial structure generation, fast DFT and MD engines, and leading MLFF methods makes Schrödinger the leading partner for MLFF generation. In this application note, we showcase the applications of QRNN technology in three different areas of materials science, namely the modeling of liquid electrolytes, polymers, and ionic liquids.
Advantages of MLFF
One limitation of classical force fields is the inability to adjust for changing partial charges and polarization of atoms, leading to inaccurate electrostatic interactions. On the other hand, MLFF methods enable the dynamic adjustment of partial charges and polarization, resulting in more accurate dynamics. Since MLFF models are trained on DFT data, they offer a higher level of accuracy that is similar to ab initio calculations. Empirical parameters-based classical force fields also breakdown when modeling interactions in complex inorganic and organic systems, while MLFF can effectively learn the underlying potential energy surface that governs these interactions. Moreover, with the use of Desmond (Schrödinger’s GPU-accelerated molecular dynamics engine), MLFF simulations can be performed at a rate of 1ns/day for a 10,000 atom system, allowing for larger systems to be simulated for sufficient periods to obtain converged bulk properties.
APPLICATION
High-Dimensional Neural Network Force Field for Liquid Electrolytes2
Liquid electrolytes are one of the most important components of Li-ion batteries, which are a critical technology of the modern world. However, it is challenging to obtain molecular-level insights into the structure of liquid electrolytes and its impact on the bulk properties. Additionally, we lack the computational tools required to quantitatively calculate key properties of the materials (viscosity, ionic diffusivity) from first principles necessary to design next-generation battery materials. The optimization of battery electrolyte formulation is a complex multi-objective problem.
Approach
In this work, we developed MLFF for liquid electrolyte simulations which bridges the gap between the accuracy of range-separated hybrid DFT and the efficiency of classical force fields. Our training dataset consists of LiPF6 salt and 7 common carbonates.* We trained the MLFF to a dataset of ~360K molecular clusters using seven rounds of active learning. The performance of the MLFF was validated by comparing the predicted values to the experimental bulk thermodynamic and transport properties over a range of temperatures and salt concentrations.
*Carbonates used in this study: ethylene carbonate (EC), propylene carbonate (PC), vinylene carbonate (VC), fluoroethylene carbonate (FEC), dimethyl carbonate (DMC), diethyl carbonate (DEC), and ethyl methyl carbonate (EMC)
Results
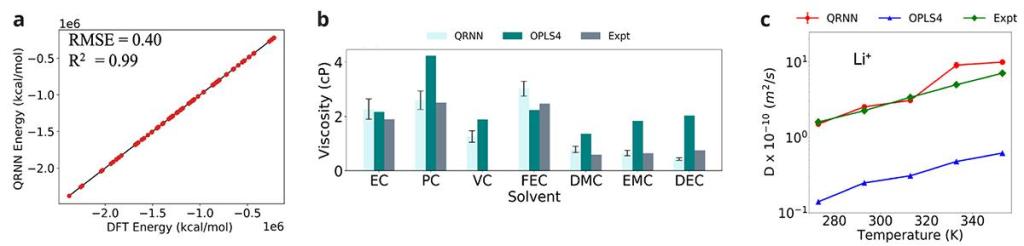
We evaluated the MLFF model performance by constructing an independently sampled test set of 2.5K cluster data points extracted from the production MD trajectories of pure carbonate solvents and electrolyte mixtures (Figure 2a). Predictions of material properties made with this force field are quantitatively accurate compared to experimental data. Such highly accurate MLFF models are critical to optimize complex electrolyte formulations needed for advancing battery technology.
- The absolute energies, dipole components, and force components are in good agreement to the reference level of DFT theory.
- The MLFF predicted bulk properties (such as density, viscosity, heat of vaporization, etc.) are in excellent agreement with experimental values, and provide significant improvement in the predicted properties for the pure liquid electrolytes as compared to the OPLS4.
- The MLFF computed diffusivities are in excellent agreement with the experimental data.
APPLICATION
Development of Scalable and Generalizable MLFF for Polymers3
Classical force fields such as OPLS have been successful for polymer systems in capturing some properties such as density at a constant temperature, however, for certain dynamic and thermodynamic properties such as self-diffusivity, viscosity, and specific heat, it is limited to primarily obtaining relative trends.
Approach
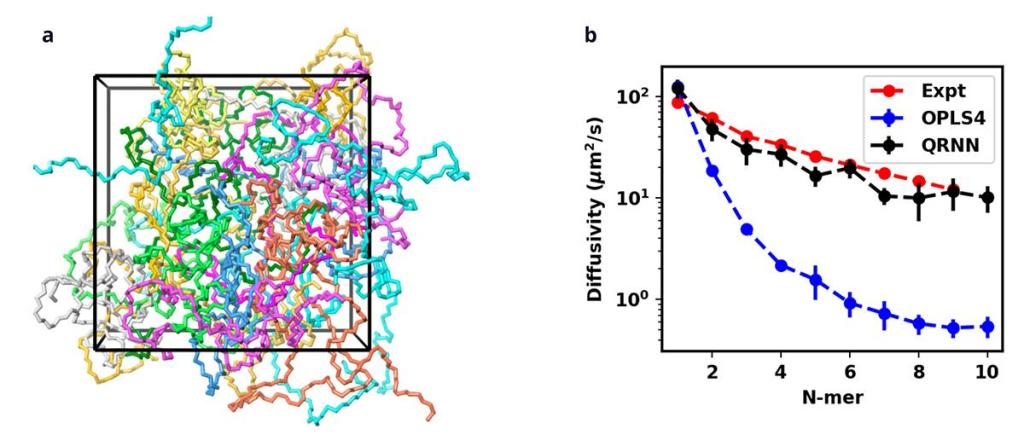
We developed an MLFF workflow for polymers and demonstrated its scope by applying it to ethylene glycol oligomers. We trained on samples containing monomers, dimers, and trimers of ethylene glycol to create an MLFF model and evaluated the model by predicting the oligomer properties (density, self-diffusivity, and specific heat) and comparing them with DFT and experimental results.
Results
We evaluated the MLFF model performance by comparing the predicted energies with the DFT energies. The below parity plot shows that the model is performing well. The MLFF potentials accurately capture the diffusion dynamics and surpass the capabilities of what the classical force field can provide (Figure 3b). This demonstrates that our MLFF for polymers is scalable and can be extended to compute complex properties dependent on the chain dynamics, such as mechanical properties and thermophysical properties, including the glass transition temperature. Our work shows that the advancing area of MLFF has application in a critically important materials area, polymers. This significant advance was not possible with previous MLFFs and is an important step toward simulations with the ideal matching of accuracy and speed for key materials design parameters.
APPLICATION
Machine Learning Fitted Force Fields for Ionic liquids
Ionic liquids represent an intermediate class of molecules that are liquid at room temperature, but are composed of completely charged cation and anions. They are useful for advanced batteries, biopharmaceuticals, and as industrial solvents. The charged nature of ionic liquids make them difficult to simulate and can only be correctly fitted with more expensive polarizable force fields. DFT based simulations are limited to only simulating 10s of molecules.
Approach
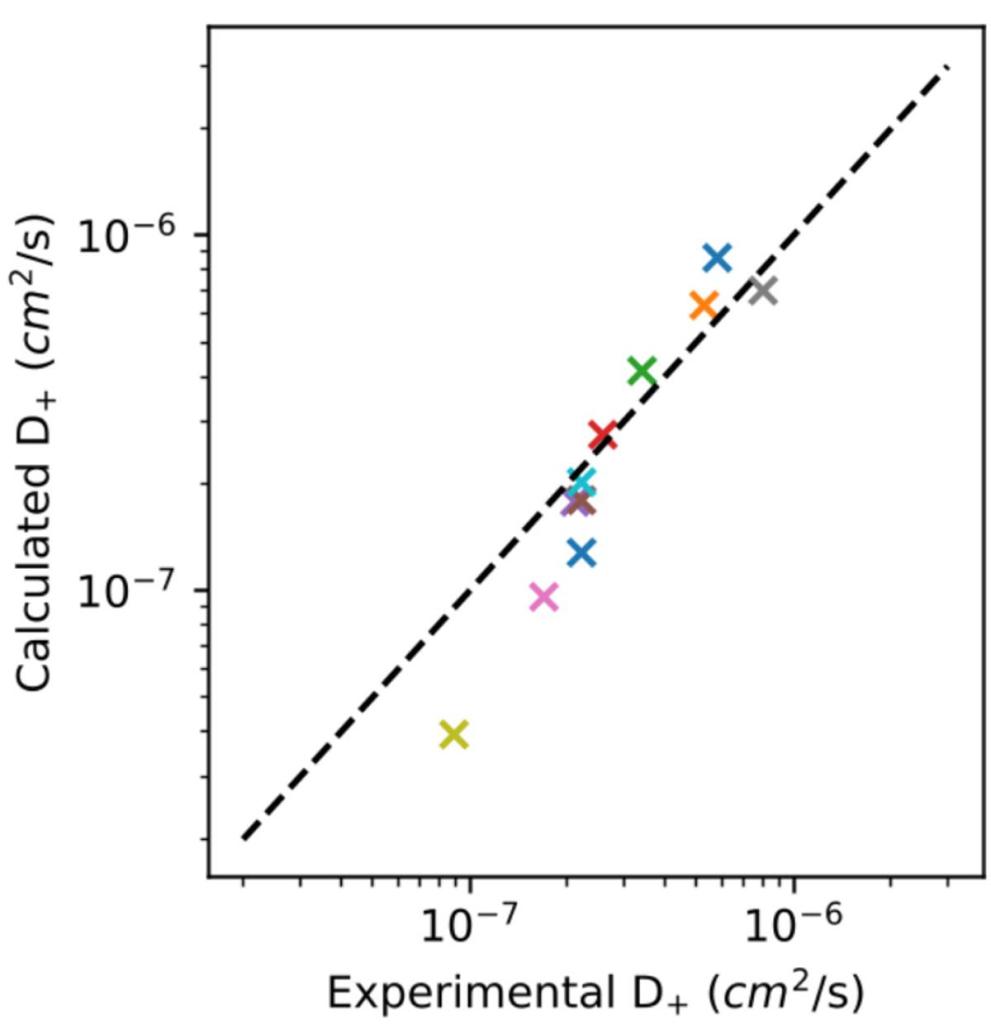
We train an ML force field on DFT energies and forces for neutral organic molecules, charged organic molecules, dimers, and small clusters. We then run molecular dynamics on a simulation box containing between 1000- 3000 atoms for several classes of ionic liquids.
Results
MLFF ensemble models enable stable simulation performance of simulations over 2ns. The diffusion coefficients predicted by the MLFF agree with experimental results over a wide range of ionic liquid chemistries (Figure 4). These results demonstrate the first ab initio based simulation of ionic liquids at such length and timescales.
How we can help
Schrödinger provides research services focused on the development of advanced machinelearned force fields to facilitate accurate molecular dynamics simulations for a range of applications and to enable modeling of complex material systems with high speed and accuracy.
References
-
Jacobson, L. D., Stevenson, J. M., Ramezanghorbani, F., Ghoreishi, D., Leswing, K., Harder, E. D., & Abel, R. (2022). Transferable neural network potential energy surfaces for closed-shell organic molecules: Extension to ions.
Journal of Chemical Theory and Computation, 18(4), 2354-2366.
-
Dajnowicz, S., Agarwal, G., Stevenson, J. M., Jacobson, L. D., Ramezanghorbani, F., Leswing, K., … & Abel, R. (2022). High-dimensional neural network potential for liquid electrolyte simulations.
The Journal of Physical Chemistry B, 126(33), 6271-6280.
-
Mohanty, S., Stevenson, J., Browning, A., Jacobson, L., Leswing, K., Halls, M., & Afzal, M. (2023). Development of Scalable and Generalizable Machine Learned Force Field for Polymers.
ChemRxiv. doi:10.26434/chemrxiv-2023- 4z2bw This content is a preprint and has not been peer-reviewed.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.