
Opening new worlds for molecular discovery
Schrödinger’s computational platform, powered by physics, is transforming the way therapeutics and materials are discovered to make innovations of the future achievable, today.

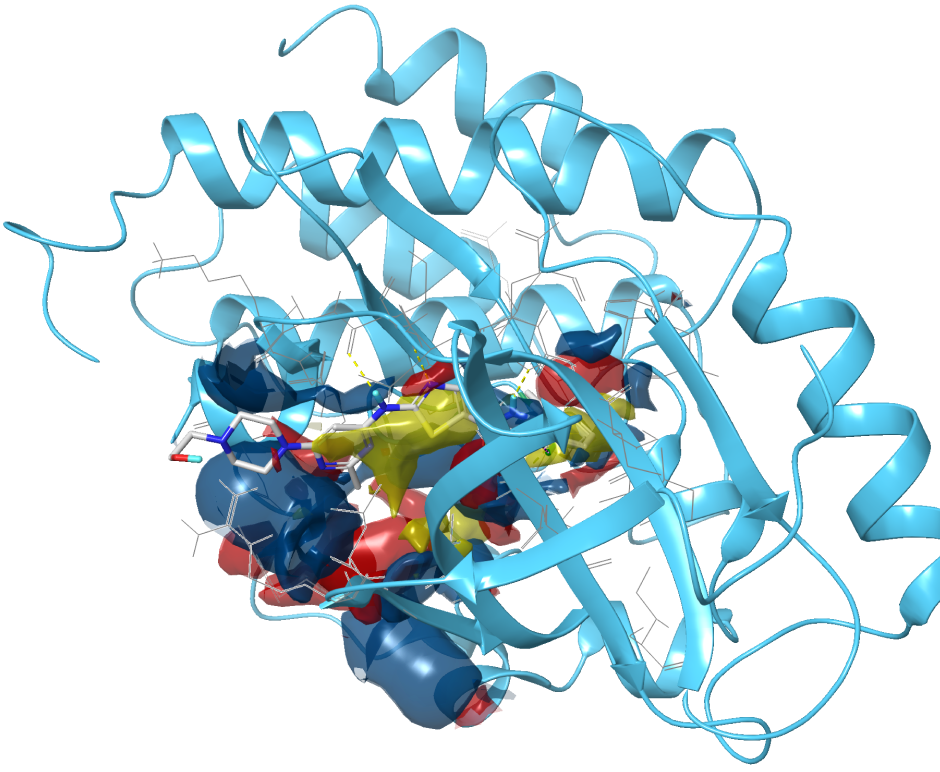
Leverage the power of our industry-leading platform to discover and optimize better molecules, faster.
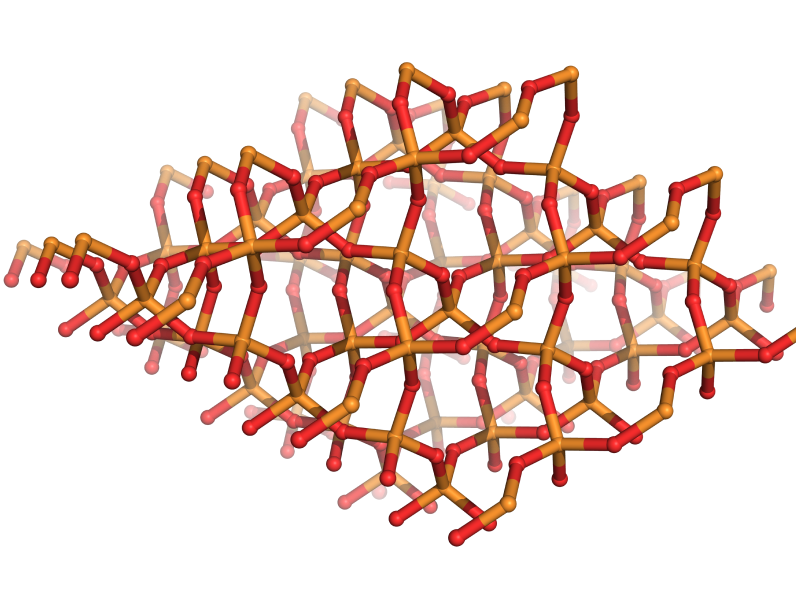
Discover, optimize, and formulate the materials of the future with our predictive computational platform.

Schrödinger’s therapeutics group strives to reduce risk in drug discovery and development by selecting high-value targets with established human genetics or clinical validation, where we can solve a design challenge, and where we believe we can deliver value for patients.
Level up your skills with educational resources developed by industry leaders.

Practical learning and hands-on application of workflows using Schrödinger technology
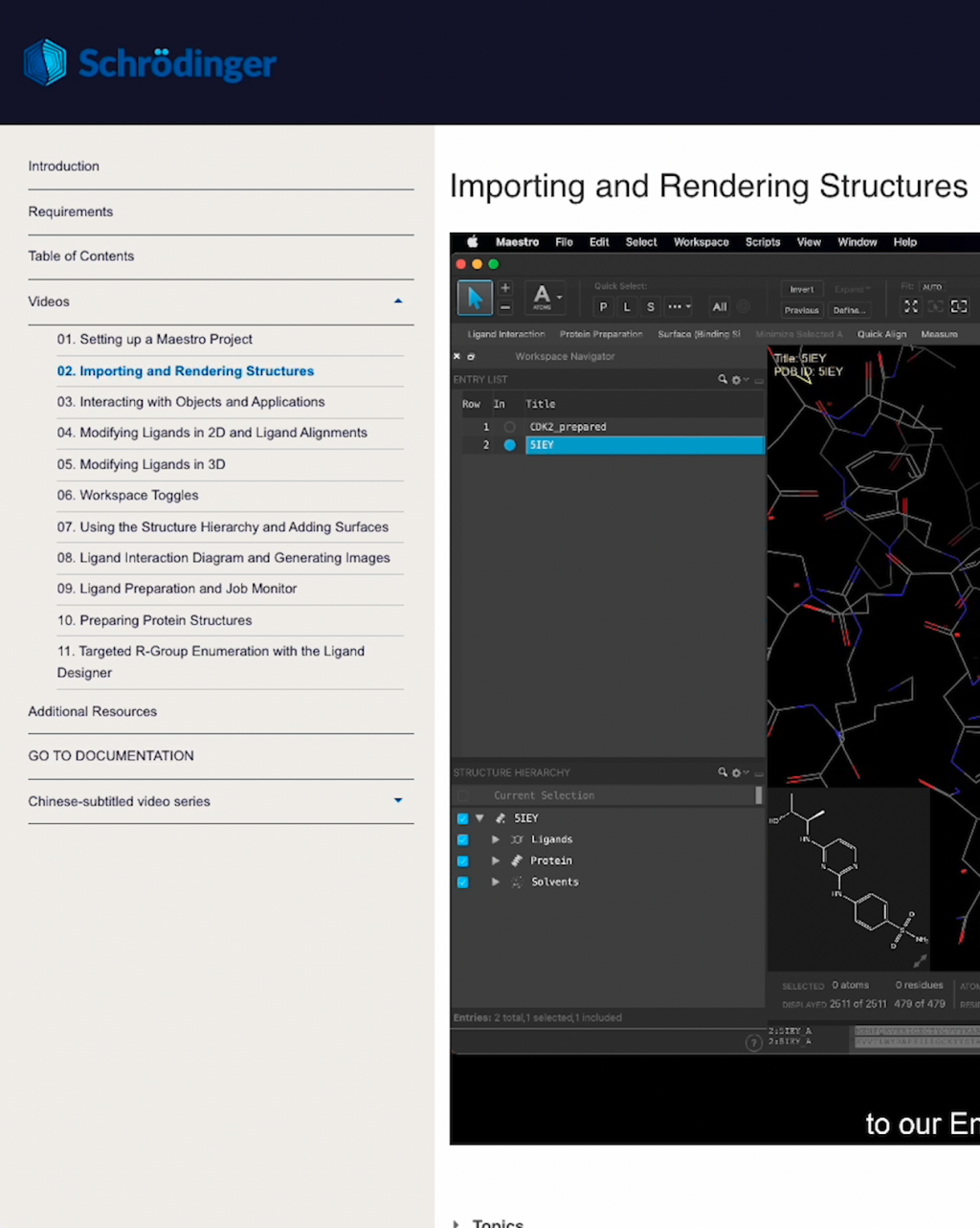
Step-by-step instructions for performing tasks and analyzing data

Industry-leading software in the classroom to learn theoretical and applied sciences







Join us for a series of live webinar presentations from leading educators at top academic institutions, as well as talks by Schrödinger scientists.