Hit to lead design of novel d-amino-acid oxidase inhibitors using a comprehensive digital chemistry strategy
Computational platform grounded in highly accurate predictive models enables team-based discovery of a novel chemical series engaging a complex CNS target.
Overview
Inhibition of D-amino-acid oxidase (DAO) has been hypothesized as a potential therapeutic strategy for schizophrenia. Schrödinger’s Drug Discovery Team engaged in a discovery effort with a collaborator to identify novel DAO inhibitors with potential best-in-class properties.
Program Challenges
- Identify novel chemical matter while striving for best-in-class molecules that cross the blood-brain-barrier
- Simultaneously optimize drug-like properties, improve CNS exposure, and affinity
Approach
The Drug Discovery Team deployed a large-scale digital chemistry strategy leveraging:
- A centralized project data platform to facilitate knowledge-based medicinal chemistry design collaboration (LiveDesign, AutoQSAR)
- Physics-based methods to predict affinity and prioritize design ideas for synthesis (FEP+)
- Computationally-driven ideation and scoring workflow to amplify common enumeration strategies and screen hundreds of millions of compounds using machine learning coupled with physics-based free energy methods (FEP+, AutoDesigner)
Results
The team discovered a novel class of DAO inhibitors with desirable drug-like properties by confidently exploring synthetically-challenging chemistry. The team also identified a previously unexplored subpocket for further evaluation. The novelty of the compounds, coupled with well-balanced properties, demonstrates the extraordinary power of the approach to unleash project team creativity. By leveraging a digital platform, the team explored vast chemical space while simultaneously optimizing for drug-like properties in a challenging disease area.
Why use a digital chemistry approach?
A digital chemistry approach uses physics-based modelling, machine learning, and a team-based collective intelligence platform to design better molecules on accelerated timelines.
How to achieve optimal drug-like properties?
The development of CNS drugs poses several unique challenges. Fine-tuning physicochemical properties for optimal brain exposure is an essential element of CNS drug development. Many companies have discontinued neuroscience discovery because these challenges lead to longer development timelines and a lower probability of success.
Schrödinger’s Drug Discovery Group developed property prediction models, using AutoQSAR deployed via LiveDesign, a web-based collaborative design platform. LiveDesign enabled teams of medicinal chemists to crowdsource designs and simultaneously optimize CNS properties with push-button workflows in a single interface (see figure 1). Compounds predicted in the desired property space were triaged using free energy perturbation (FEP+), a physics-based method for accurately predicting compound binding affinity. This workflow empowers teams to confidently pursue synthetically challenging compounds.
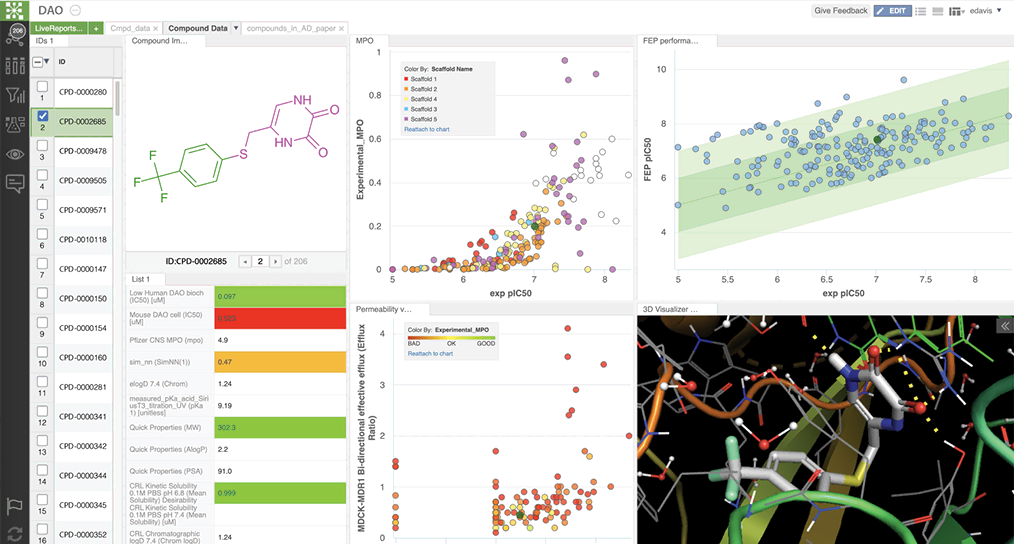
How did a digital chemistry strategy enable improved hypothesis testing?
The team delivered high-quality molecules by working in an ecosystem that facilitated exploration of vast, novel chemical space and simultaneous optimization for desired properties through accurate physics-based modeling and machine learning.
The digital chemistry approach allowed the team to discover and quickly overcome many medicinal chemistry challenges in the pursuit of best-in-class molecules (see figure 1).2

The team interrogated the atypical polarity of the DAO binding site, shown in panel A of figure 3. Chemists pursued challenging chemistry to reduce conformational flexibility and displace a high-energy water molecule by cyclizing and methylating cmpd 4 and 5 (see figure 2, panels B and C).
Finally, while literature and crystallographic structures suggested limited pocket volume for SAR exploration,3 FEP+ revealed the opportunity to interrogate this vector with larger chemical groups such as cmpd 6, as shown in panel D of figure 3.

How to interrogate vast chemical space and deliver novel chemical matter?
Pursuing best-in-class molecules required exploring vast chemical space outside of previously characterized drug-like molecules. The team utilized AutoDesigner, a multifaceted large-scale enumeration workflow (figure 4), to generate ideas exploring the novel DAO binding subpocket suggested by FEP+ (see figure 3, panel D).4
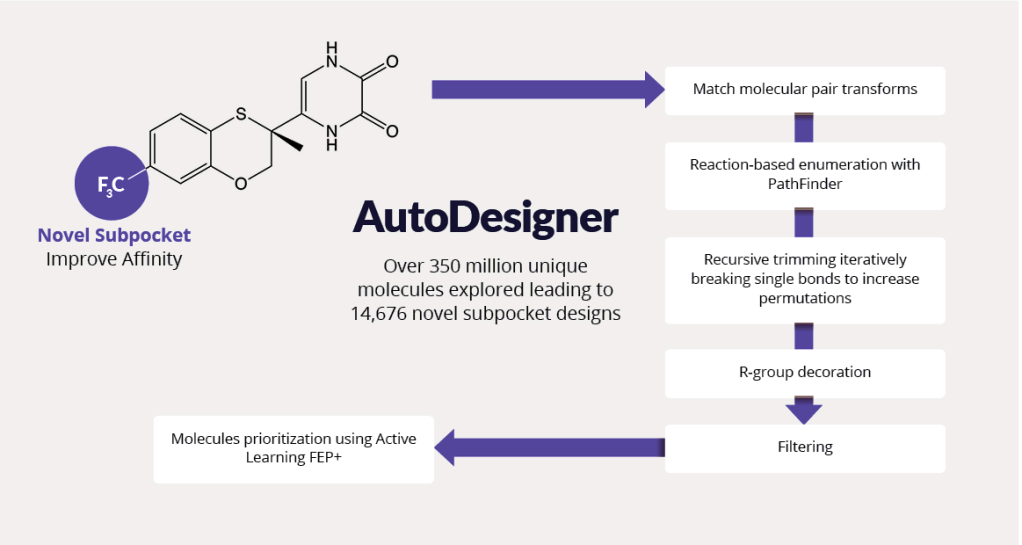
To explore SAR and tune physicochemical properties, the team performed iterative cycles of AutoDesigner in the newly discovered subpocket. After triaging with appropriate filters, all molecular ideas were prioritized using free energy methods. The team trained active learning models using physics-based affinity predictions (FEP+) to prioritize compounds for synthesis. In total, more than 350 million ideas were generated and triaged.
What was the project impact?
Typically as drug discovery programs progress, teams struggle to balance desired properties, which leads to deficits in desired drug properties as novel scaffolds are explored and optimized.
Through a computational platform rooted in creative team collaboration, highly accurate predictive modeling, and enhanced by machine learning, a promising CNS DAO inhibitor series transitioned from hit discovery to lead optimization with approximately 11,000 compounds scored by FEP+ and only 208 synthesized. Of the 208 compounds synthesized, only 20 were inactive (>10μM) against DAO. By discovering novel compounds and concurrently performing multi-parameter optimization of critical CNS properties, the team delivered high project impact for this challenging disease area.
References
- Bromet E.J., Fenning S.; Epidemiology and natural history of schizophrenia. Biol Psychiatry. 1999, 46 (7), 871–881.
- Tang et al. Discovery of a Novel Class of D-amino Acid Oxidase (DAO) Inhibitors with the Schrödinger Computational Platform. ChemRxiv. Preprint. https://doi.org/10.33774/chemrxiv-2021-dkf1k.
- Hondo et. al. 4-Hydroxypyridazin-3(2H)-one Derivatives as Novel d-Amino Acid Oxidase Inhibitors. J Med Chem. 2013, 56 (9): 3582-3592.
- Bos et al. AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization: Application to the Design and Synthesis of D-Amino Acid Oxidase Inhibitors. ChemRxiv. Preprint.