

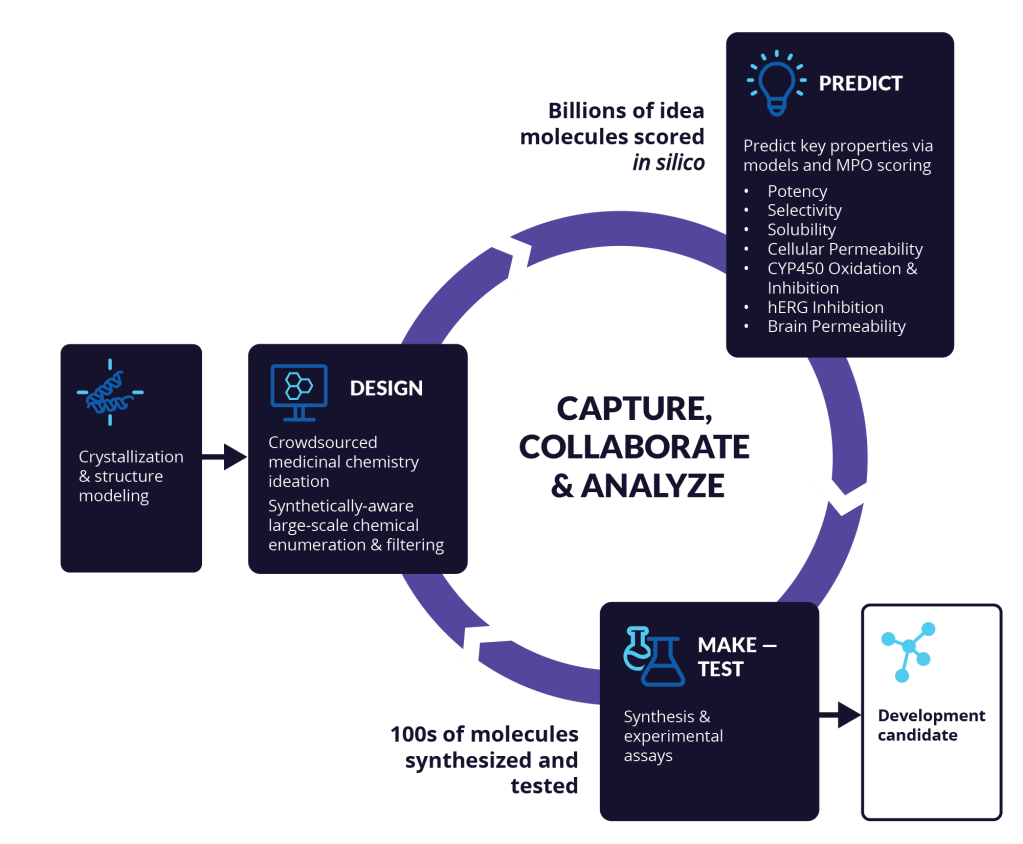
Capture your best ideas and drive more productive design cycles
As a medicinal chemist, you know that efficiency matters. Whether it’s finding ways to quickly test a hypothesis or rapidly iterate on an idea, Schrödinger’s computation platform for drug discovery allows you to exponentially expand the pool of hypotheses that can be tested in silico.
Use highly accurate digital assays to confidently spend your time and energy exploring vast, diverse chemistry and send only the top performing molecules for synthesis. Leverage a centralized platform for molecular design to share ideas and results across project teams in real-time to accelerate learning and collaboration in pursuit of the best quality molecules.

Integrated solution for molecular design and discovery
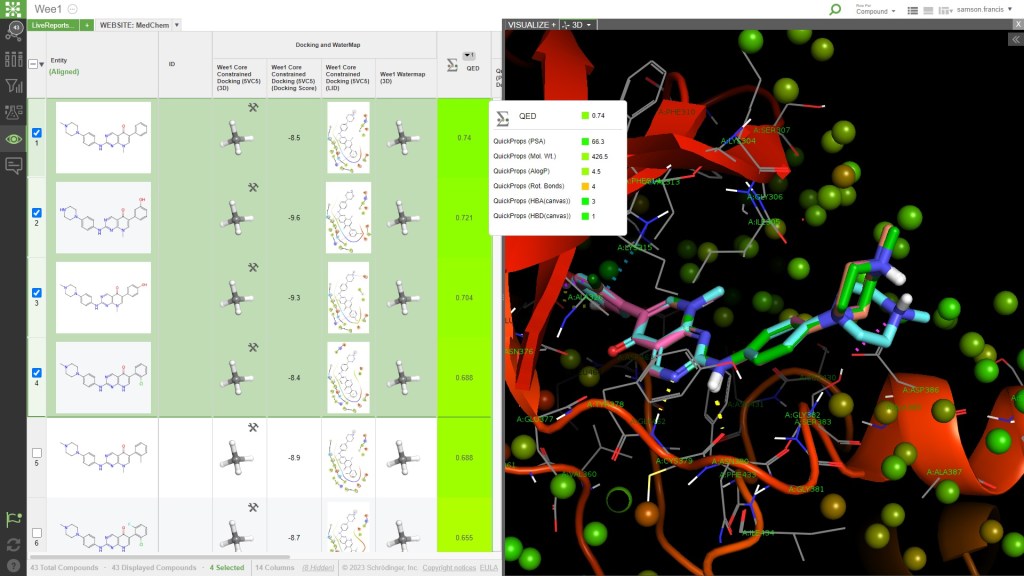
Design, predict, analyze, and collaborate in a single enterprise platform
- Design and test ideas using powerful predictive modeling workflows at your fingertips
- Crowdsource ideas and interactively revise design strategies with your colleagues – anytime, anywhere in LiveDesign
- Build rich dashboards to analyze entire datasets or individual molecules in real-time
- Track compound progression with internal and CRO partners using live data updates
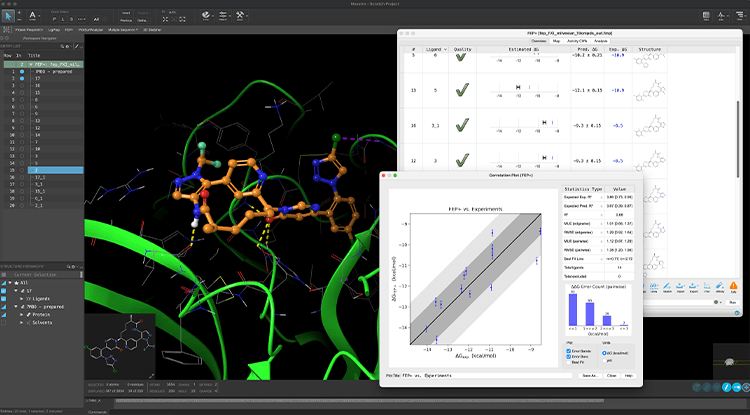
Confidently prioritize compounds for synthesis using a highly accurate in silico assay
- Predict protein-ligand binding affinity for both on- and off-targets across broad chemical space at an accuracy approaching experimental methods with FEP+
- Predict amorphous and crystalline solubility, membrane permeability, and CNS penetration with rigorous physics-based methods
- Model off-target binding modes to inform the reduction of liabilities including hERG and CYPs
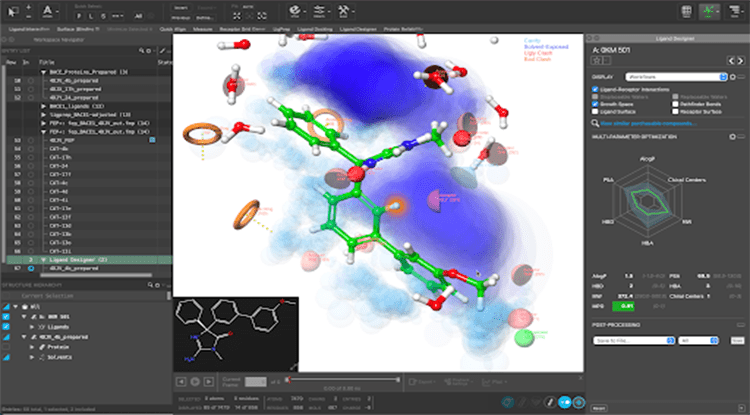
Perform intuitive, interactive 3D visualization and molecular design
- Design ligand modifications in both 2D or 3D and visualize which changes are likely to impact protein-ligand complex structures with Ligand Designer
- Rationalize SAR, drive potency, and tune selectivity by leveraging information about the entropy and enthalpy of hydration sites using WaterMap
You’re in good company
“Seeing the ways in which different people on your team think can really open up new possibilities. We are often in meetings with LiveDesign open in front of us, editing in real time, presenting ideas, and making decisions about the best designs. This makes it seamless to create a report in real-time that everyone on the team has contributed to.”

“For the program I lead, we leveraged accurate physics-based predictive models to explore a breadth of chemical space far wider than what one would survey traditionally. This allowed the team to move with focus and speed through the hit ID stage, identifying multiple attractive, novel, developable starting points. We can now hone in on the chemical matter most likely to bring us success, whilst having a number of backup options at our disposal. This puts us in a great position as we advance the program forward at an accelerated pace.”


Our platform in action


Solutions for all stages of your drug discovery program
Get more from your ideas by harnessing the power of large-scale chemical exploration and accurate in silico molecular prediction.
Structure prediction & target enablement
Hit discovery
Hit-to-lead & lead optimization
Drug formulation
Case studies & webinars
Hit to development candidate in 10 months: Rapid discovery of a novel, potent MALT1 inhibitor
Design of a highly selective, allosteric, picomolar TYK2 inhibitor in clinical development
The predict-first paradigm: How digital chemistry is changing drug discovery
Learn advanced molecular modeling tools at your own pace
 Life Science
Life Science
Life Science
Life Science
Introduction to molecular modeling in drug discovery
Protein preparation, ligand docking, collaborative design, and other fundamentals of small molecule drug discovery with Maestro and LiveDesign
 Life Science
Life Science
Life Science
Life Science
Free energy calculations for drug design with FEP+
Running, analyzing, and troubleshooting relative binding FEP+ calculations for small molecule lead optimization
Software and services to meet your organizational needs
Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Services
Leverage Schrödinger’s computational expertise and technology at scale to advance your projects through key stages in the drug discovery process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.