Our Platform
Designing the molecules of tomorrow
demands a ‘predict-first’ approach

Designing the molecules of tomorrow
demands a ‘predict-first’ approach
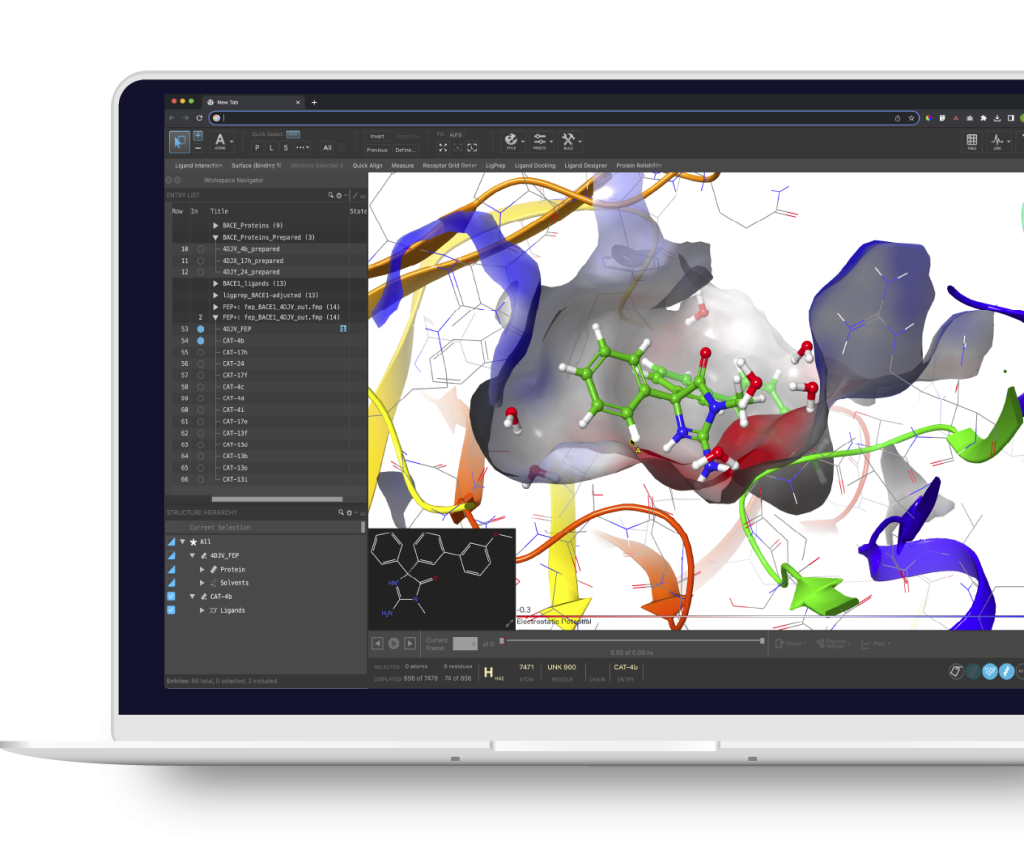


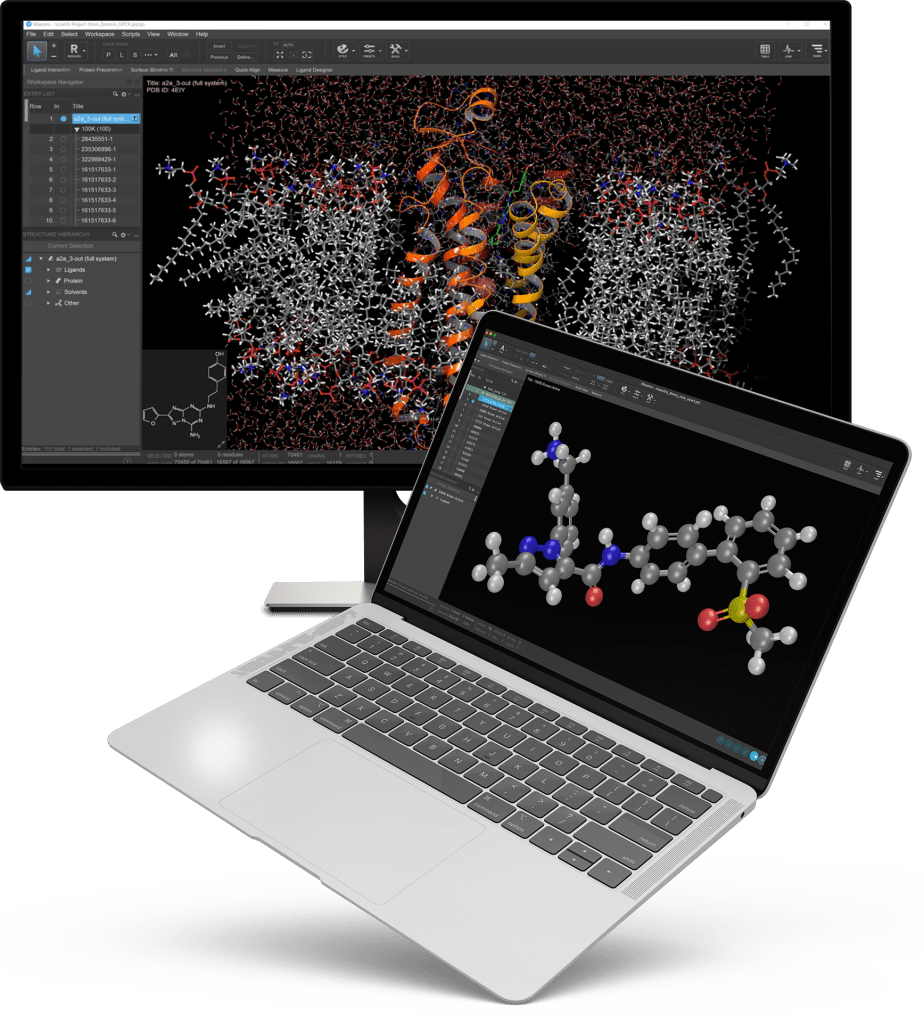
Accelerating the convergence of physics, machine learning and enterprise informatics
Built upon more than 30 years of R&D, our industry-leading computational platform is transforming the way therapeutics and materials are discovered by enabling highly accurate in silico predictions of key molecular properties across vast chemical space.



High-precision design of novel therapeutics through exploration of vast chemical space and accurate prediction of key properties of molecules

The industry-leading software platform for discovery and optimization of molecules for today’s biotech and pharmaceutical innovators

A pipeline of collaborative and proprietary drug discovery programs, powered by our platform

Design higher-performing, more sustainable materials while accelerating development timelines and gaining an improved understanding of development processes.
The power of our platform lies in a continuous feedback loop. We invest heavily in the science underlying our technology, and we use our platform internally to advance a pipeline of proprietary and collaborative therapeutics.
The insights we gain are fed back into the software, further refining its predictive capabilities and expanding the breadth of applications it supports. This ongoing innovation is delivered back to our users through regular software updates.
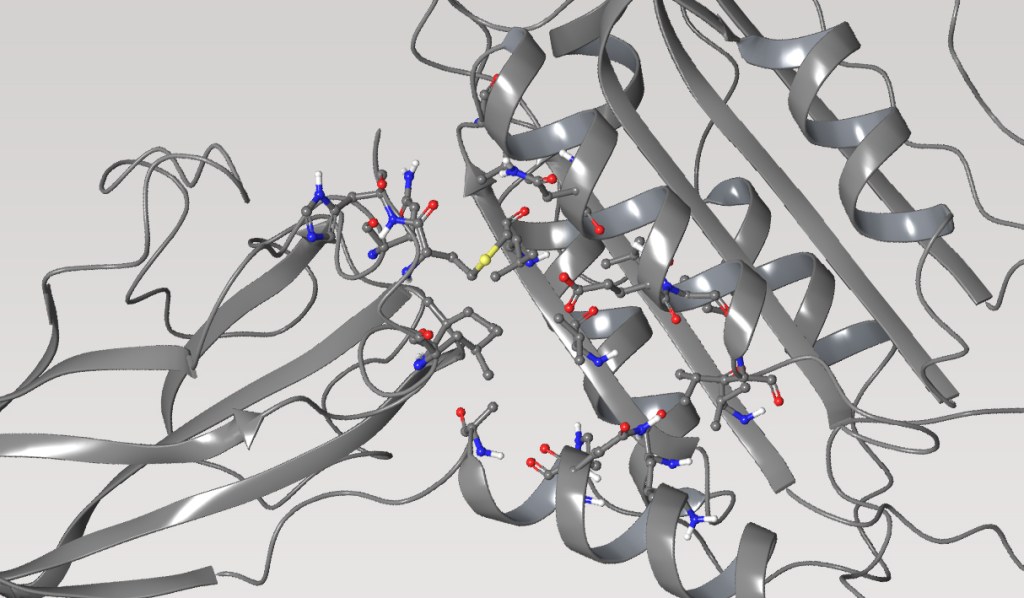 Case Study
Case Study
 Case Study
Case Study