BioLuminate
Comprehensive modeling platform for biologics discovery

Comprehensive modeling platform for biologics discovery
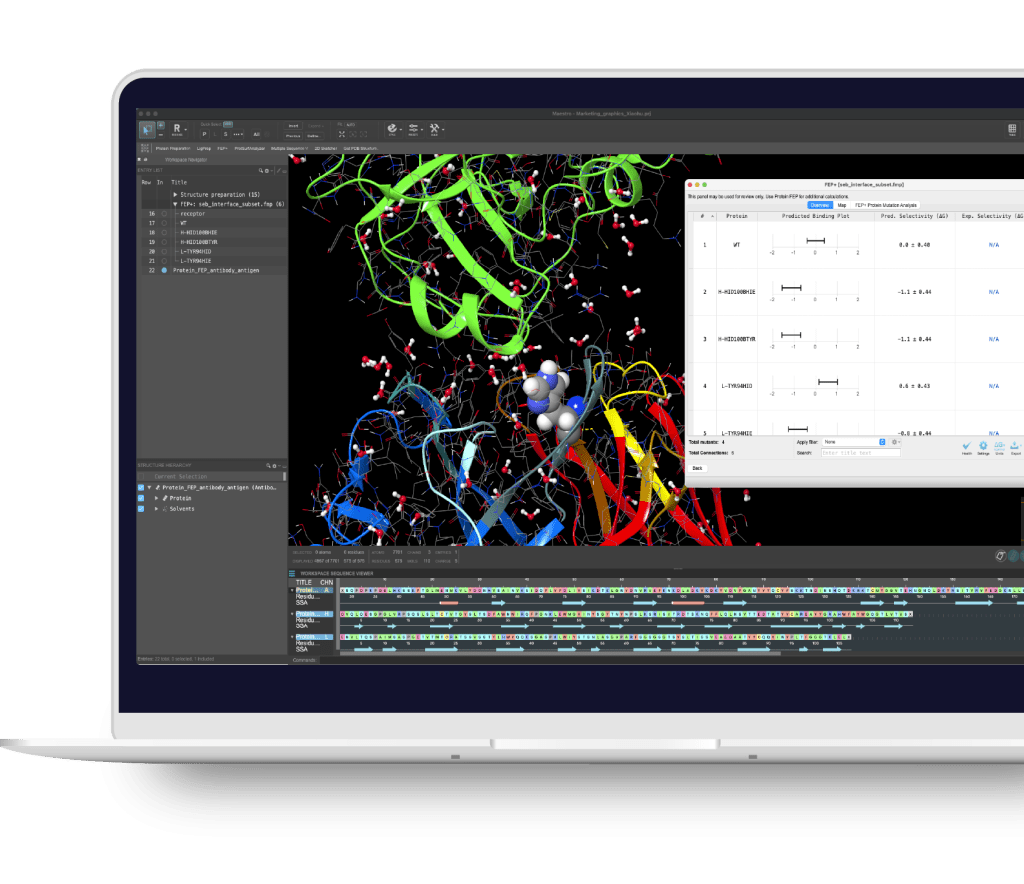
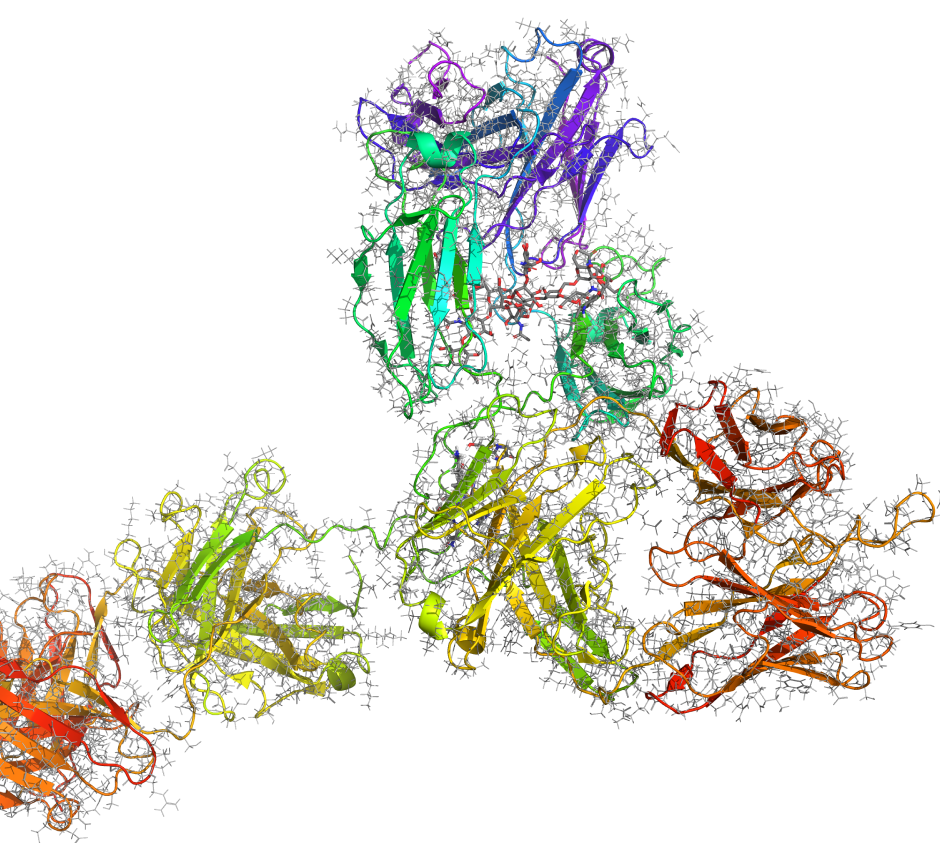
Maestro BioLuminate is Schrödinger’s comprehensive interface to streamline computationally-guided biologics drug discovery. By leveraging industry-leading molecular simulations and logically organizing tasks and workflows, BioLuminate provides predictive methods to optimize properties of biomolecules from sequence to structure.

Access refined workflows across multiple biological modalities
Get answers to common questions and learn best practices for using Schrödinger’s software.

Level-up your computational modeling skills and enroll in our online course, Introduction to Computational Antibody Engineering.
view courseDiscover how Schrödinger technology is being used to solve real-world research challenges.
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.