Docking and scoring
Virtual screening has proven to be a very successful approach for finding ligand hits and assisting lead optimization in structure-based drug discovery projects. By docking a large library of compounds into one or more high-resolution structures of the target receptor, fewer compounds typically need to be experimentally screened to identify prospective lead optimization candidates. Beyond identifying small molecules likely to bind well to a protein target, docking methods are used in a variety of context such as polypeptide and macrocycle pose prediction, predicting protein-ligand complex geometries, and preparing congeneric series for binding affinity prediction with methods such as Free Energy Perturbation or MM-GBSA. Moving beyond the rigid receptor approximation common in structure-based virtual screening, the Induced Fit docking protocol predicts the effect of ligand docking on protein structure.
Rigid Receptor Docking with Glide
Glide Docking and Scoring Methodology
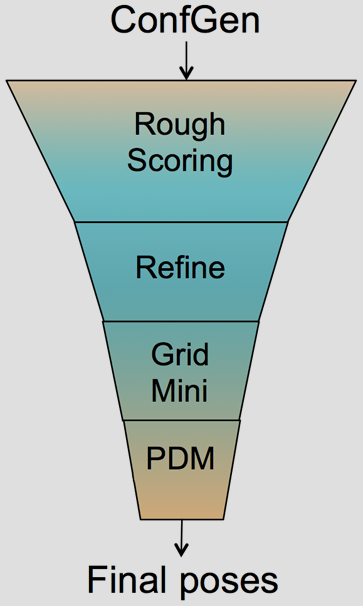
The Glide HTVS, SP and XP docking methodologies have previously been described in detail.1-3 Glide HTVS and SP use a series of hierarchical filters to search for possible locations of the ligand in the binding-site region of a receptor. The shape and properties of the receptor are represented on a grid by different sets of fields that provide progressively more accurate scoring of the ligand pose. Exhaustive enumeration of ligand torsions generates a collection of ligand conformations that are examined during the docking process. Given these ligand conformations, initial screens are deterministically performed over the entire phase space available to the ligand to locate promising ligand poses. From poses selected by initial screening, the ligand is refined in torsional space in the field of the receptor using OPLS34 (Glide SP & XP) or OPLS2005 (GLIDE HTVS) with a distance-dependent dielectric model. Finally, a small number of poses are minimized within the field of the receptor with full ligand flexibility (post-docking minimization or PDM). This process is referred to as the docking funnel as illustrated in Figure 1.
Glide HTVS can dock compounds at a rate of about 2 seconds/compound and trades sampling breath for higher speeds. Glide SP performs exhaustive sampling and is the recommended balance between speed and accuracy, requiring about 10 seconds/compound. Glide XP employs an anchor-and-grow sampling approach and a different functional form for GlideScore. It can dock compounds at a rate of about 2 minutes/compound. These three docking modes provide an array of options in the balance of speed vs. accuracy for most situations.
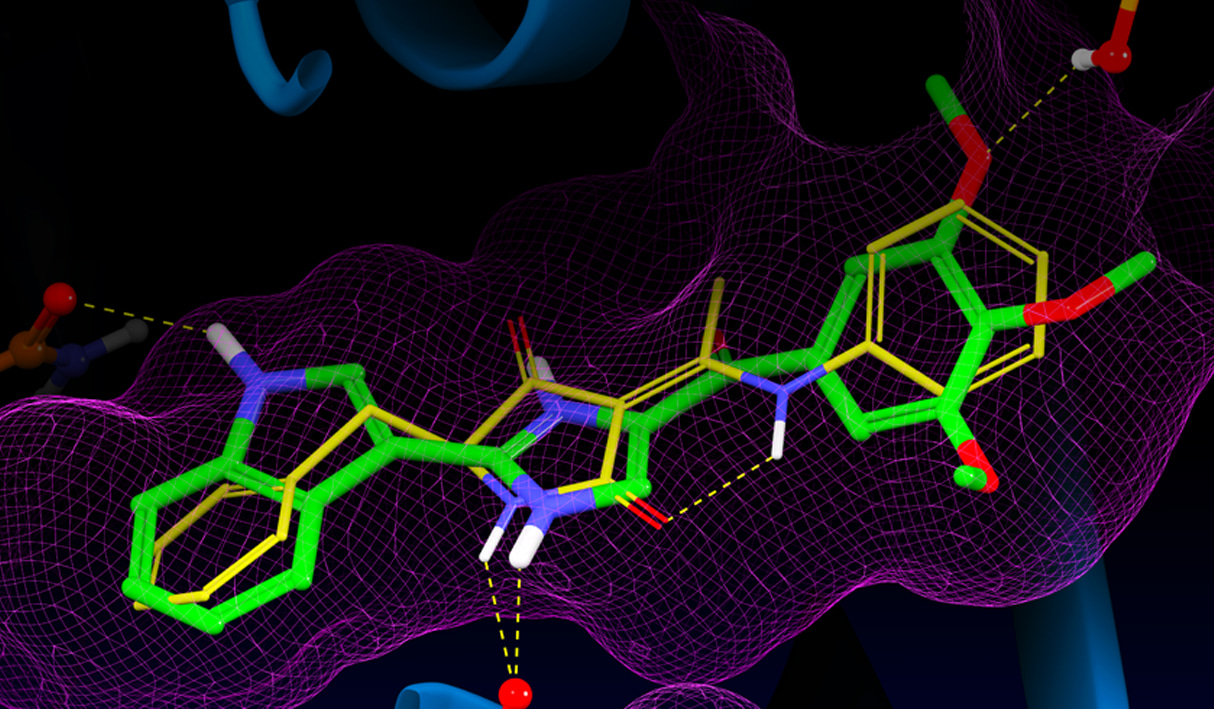
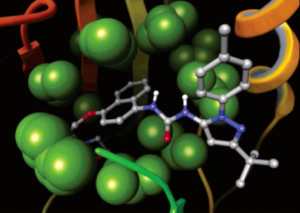
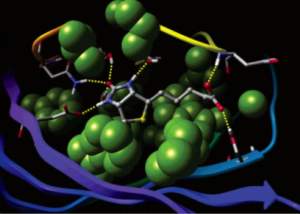
Glide uses the Emodel1 scoring function to select between protein-ligand complexes of a given ligand and the GlideScore2 function to rank-order compounds to separate compounds that bind strongly (actives) from those that don’t (inactives). The Emodel scoring function is primarily defined by the protein-ligand coulomb-vdW energy with a small contribution from GlideScore. GlideScore is an empirical scoring function designed to maximize separation of compounds with strong binding affinity from those with little to no binding ability. As an empirical scoring function it is comprised of terms that account for the physics of the binding process including a lipophilic-lipophilic term, hydrogen bond terms, a rotatable bond penalty, and contributions from protein-ligand coulomb-vdW energies. In addition to these terms, GlideScore includes terms to account for hydrophobic enclosure3, which is the displacement of water molecules by a ligand from areas with many proximal lipophilic protein atoms. Particularly beneficial to binding is the formation of one or more protein-ligand hydrogen bonds within regions of hydrophobic enclosure as illustrated in Figures 2 and 3.
Glide Docking Accuracy and Enrichment Performance
The first Glide article1 is the 2nd most cited paper ever in the Journal of Medicinal Chemistry (as of Feb. 2016) with many references citing its application in drug discovery projects. Glide’s ability to consistently identify hits for lead optimization and guide understanding of the key interactions and desolvation effects impacting protein-ligand binding have helped drive its widespread adoption. A pre-requisite to obtaining the highest-quality docking results is to use protein and ligand structures prepared using best-practices methods by the Protein Preparation Wizard and LigPrep. As part of an unbiased comparison of docking methods, we have evaluated Glide SP for binding mode prediction using the Astex5 set of self-docking tests, and enrichment in a virtual screening context using the DUD dataset6. The datasets, evaluation metrics, and experiments performed were prescribed by conditions of the Docking and Scoring Symposium at the 241st ACS Meeting in Anaheim.7
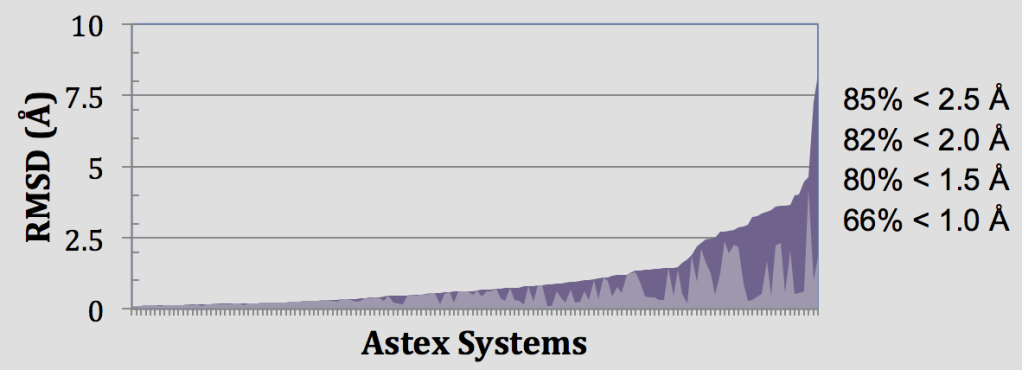
In these retrospective experiments, Glide SP reproduces crystal complex geometries in 85% of Astex cases with < 2.5 Å RMSD as shown in Figure 4. Glide is also effective in enriching screened databases in known actives, beating random selection in 97% of DUD targets and giving an average AUC of 0.80 across all 39 target systems. Early enrichment performance shows on average 12%, 25%, and 34% of known actives are recovered in screening the top-ranked 0.1%, 1%, and 2% of recovered decoys. This enrichment is particularly impressive given the structural similarity between active and decoy ligands in the DUD dataset8 and that docking a ligand with SP Glide takes only about 10 seconds using modern hardware.
| ROC(0.1%) | ROC(1%) | ROC(2%) | AUC | |
|---|---|---|---|---|
| Best-Practices Structures | ||||
| Average | 0.12 | 0.25 | 0.34 | 0.80 |
| Median | 0.06 | 0.19 | 0.27 | 0.14 |
| Std. Dev. | 0.17 | 0.22 | 0.24 | 0.14 |
| Maximum | 0.82 | 0.88 | 0.88 | 0.98 |
Using Glide to “Stay Close to Experiment”
Glide’s wide range of constraints enable one to “stay close to experiment“ which has been reported as a common theme in pharmaceutical projects at Roche that were positively impacted by computational chemistry.9 Hydrogen bond constraints require formation of a hydrogen bond to particular functional groups on the receptor, positional constraints can enforce docked poses to place particular chemistry in a volume of space relative to the receptor, metal constraints enforce coordination geometries. Core constraints are particularly valuable for enforcing expected binding modes of congeneric series in preparing docked poses for binding affinity prediction via MM-GBSA or Free Energy Perturbation.
Docking Macrocycle
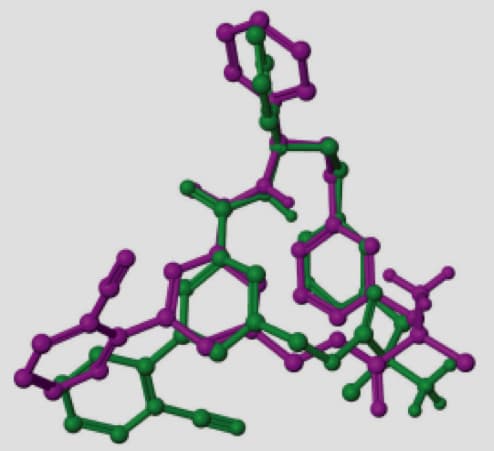
Efficient sampling of ring conformations is essential to reproduce experimental binding modes in docking experiments, particularly for macrocycles where there can be hundreds of low energy conformations. Glide relies on an extensive database of ring conformations to sample low-energy states and otherwise does not sample macrocycle ring systems. Generating ring templates for macrocycles considerably improves native pose prediction.
A representative example is the macrocycle from 2QKZ shown in this figure where the native ligand buries the macrocycle ring into a mostly enclosed pocket. To bind in this pocket requires a particular ring conformation that forms key interactions to a ligand carbonyl and protonated amine moieties. Using ring templates for this macrocycle the docked pose has a low RMSD of 0.22 Å. Without the macrocycle ring templates, even with a visually similar ring conformation of RMSD 0.71 Å for macrocycle ring atoms, Glide positions the ring outside the pocket leading to a very large RMSD of 10.23 Å.
Docking Polypeptides
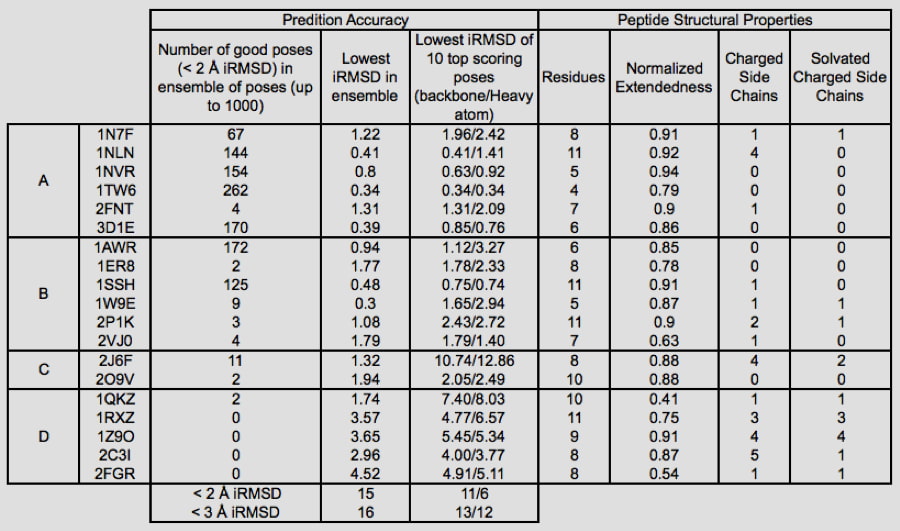
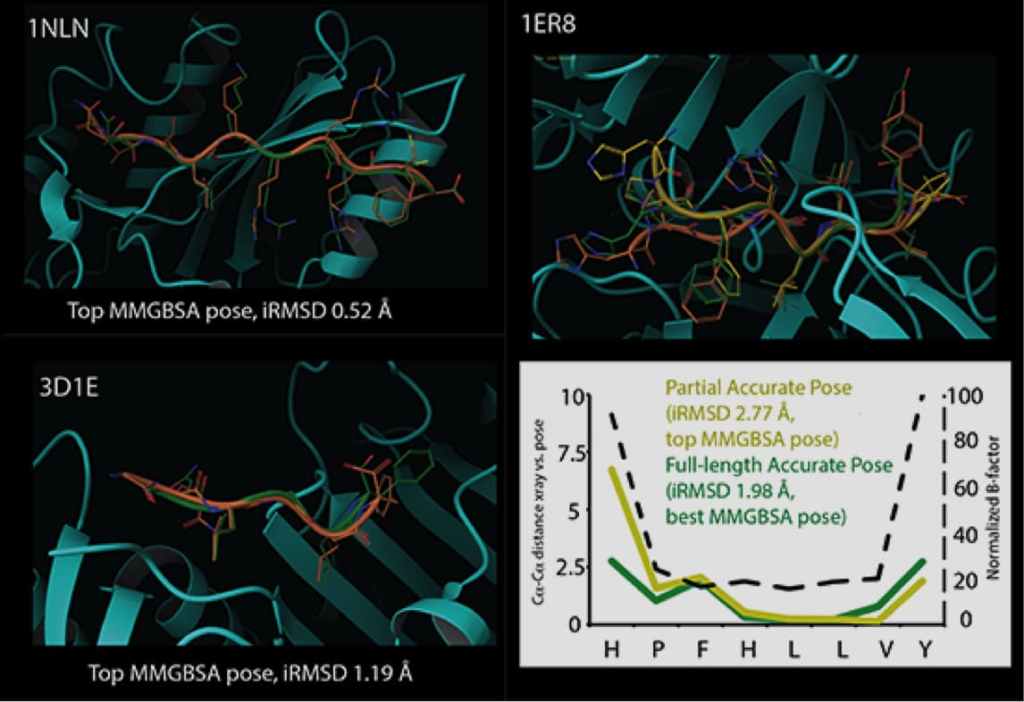
The increased use of peptides and peptidomimetics as drugs has created a need for the accurate prediction of binding complex geometries of polypeptides to receptors. The Glide SP-peptide mode10 was developed to address this need as default application of Glide to such ligands results in a successful docking for 24% of the peptides in a benchmark set. To improve binding mode prediction, the Glide sampling algorithm was modified by systematically changing all parameters that control how peptide conformations make it through the Glide funnel. Applying the optimal combination of these parameters increased pose prediction accuracy to 41%. In addition, we leveraged the previously reported sensitivity of Glide to input conformations11 and increased sampling by running multiple independent Glide jobs and pooling the results. This increased accuracy of pose prediction to 53%. Finally, by applying MM-GBSA scoring to each of the predicted poses, further boosting the accuracy to 58%. This approaches accuracy of brute-force methods like FlexPepDock (63% for this dataset) at a fraction of the computational cost (24 CPU hours for the Glide protocol).
Which Peptides are Handled Best?
Glide can dock ligands with up to 300 atoms and 50 rotatable bonds. Since many peptide ligands may exceed these limits, this effectively limits docking application to peptides of around 11 residues or less. In some cases, it is necessary to freeze the amide bonds in their trans configuration to dock peptides of longer lengths. Strong correlations are found between structural features of peptides and their ability to be docked accurately. For example, we found that reduced peptide length, increased extendedness, and small number of ionizable groups increased the probability that a peptide was docked correctly. In addition, we noted a strong correlation between the quality of prediction for segments of the peptides with low B-factors, while the high B-factor (typically the N- and/or C- termini) would have significant deviation from the crystal structure, suggesting that these terminal predictions might actually be accessible states in solution.
Induced Fit Docking
The active site geometry of a protein complex depends heavily upon conformational changes induced by the bound ligand. However, resolving the crystallographic structure of a protein-ligand complex requires a substantial investment of time, and is frequently infeasible or impossible. Schrödinger’s Induced Fit (IFD) protocol solves this problem by using Glide and Prime to exhaustively consider possible binding modes and the associated conformational changes within receptor active sites. The unique procedure allows chemists to quickly predict active site geometries with minimal expense, even for systems as challenging as hERG homology models.
The Induced Fit protocol begins by docking the active ligand with Glide. In order to generate a diverse ensemble of ligand poses, the procedure uses reduced van der Waals radii and an increased Coulomb-vdW cutoff, and can temporarily remove highly flexible side chains during the docking step. For each pose, a Prime structure prediction is then used to accommodate the ligand by reorienting nearby side chains. These residues and the ligand are then minimized. Finally, each ligand is re docked into its corresponding low energy protein structures and the resulting complexes are ranked according to a scoring function combining GlideScore and Prime energies.
The Induced Fit methodology has been thoroughly refined in real-world research applications, and is readily used by novice and expert modelers alike. As shown in Table 3 binding pose prediction of Induced Fit for a range of targets where protein conformational changes are necessary for binding is very good. In addition to default settings suitable for a wide range of systems, the Induced Fit interface features advanced options that can be customized to solve more challenging cases. Calculations can be completed in a matter of hours on a desktop machine, or in as few as 30 minutes when distributed across multiple processors.
| RMSD of Top-Ranked Poses Returned by Induced Fit
|
||||
|---|---|---|---|---|
| Target | Receptor | PDB source of ligand structure | Docking RMSD before Induced Fit (Å) |
Ligand RMSD after Induced,Fit (Å)
|
| Aldose reductase | 2acr | 1ah3 | 6.5 | 0.9 |
| Antibody DB3 | 1dba | 1dbb | 7.6 | 0.3 |
| CDK2 | 1buh | 1dm2 | 6.4 | 1.1 |
| CDK2 | 1dm2 | 1aq1 | 6.2 | 0.8 |
| CDK2 | 1aq1 | 1dm2 | 0.6 | 0.8 |
| COX-2 | 3pgh | 1cx2 | 11.1 | 1 |
| COX-2 | 1cx2 | 3pgh | 6.6 | 1.0 (0.51) |
| Estrogen receptor | 1err | 3ert | 5.3 | 1 |
| Estrogen receptor | 3ert | 1err | 2.3 | 1.4 (1.02) |
| Factor Xa | 1ksn | 1xka | 9.3 | 1.5 |
| Factor Xa2 | 1xka | 1ksn | 5.3 | 1.5 |
| HIV-RT | 1rth | 1c1c | 2.5 | 1.3 |
| HIV-RT | 1c1c | 1rth | 12 | 2.5 |
| Neuraminidase | 1nsc | 1a4q | 3.9 | 0.8 |
| Neuraminidase | 1a4q | 1nsc | 1 | 1.7 |
| PPAR – gamma | 1fm9 | 2prg | 9.1 | 1.8 (0.43) |
| PPAR – gamma | 2prg | 1fm9 | 9.8 | 3.0 (1.54) |
| Thermolysin | 1kr6 | 1kjo | 1.1 | 1.3 |
| Thermolysin | 1kjo | 1kr6 | 3.5 | 3.2 (1.65) |
| Thymidine Kinase | 1kim | 1ki4 | 4.7 | 0.4 |
| Thymidine Kinase | 1ki4 | 1kim | 0.5 | 1.2 |
4 RMSD excluding 13 atoms in the partially solvent exposed methylphenyloxazole tail of the ligand
5 RMSD excluding 6 atoms in the symmetric di-carboxylate that are flipped 180° in the IFD structureThe Receptor column lists the rigid receptors into which the ligands of other co-crystallized structures are inducing conformational changes. Prior to treatment with Induced Fit, docking results to the rigid receptor return either no poses or high RMSDs. Ligand poses improve dramatically as Induced Fit accurately predicts the active site geometry.
Covalent Ligand Docking
In recent years, there has been a growing interest in the design of drugs forming a covalent bond with the target protein, with nearly 30% of the marketed drugs targeting enzymes known to act by covalent inhibition12. Covalent interaction with the target protein has the benefit of prolonged duration of the biological effect and potential for improved selectivity. Examples of proteins that have been targeted by covalent drugs include serine penicillin-binding proteins (PBPs) which bind to β lactams and β-lactone antibiotics, cysteine proteases such as Cathepsin B, K, and S, which are covalently modified by vinyl sulfones, epoxides, isothiazolones, and ketoamides, as well as other chemical groups, and hepatitis C virus (HCV) protease, which covalently binds the ketoamide groups of boceprevir and telaprevir.
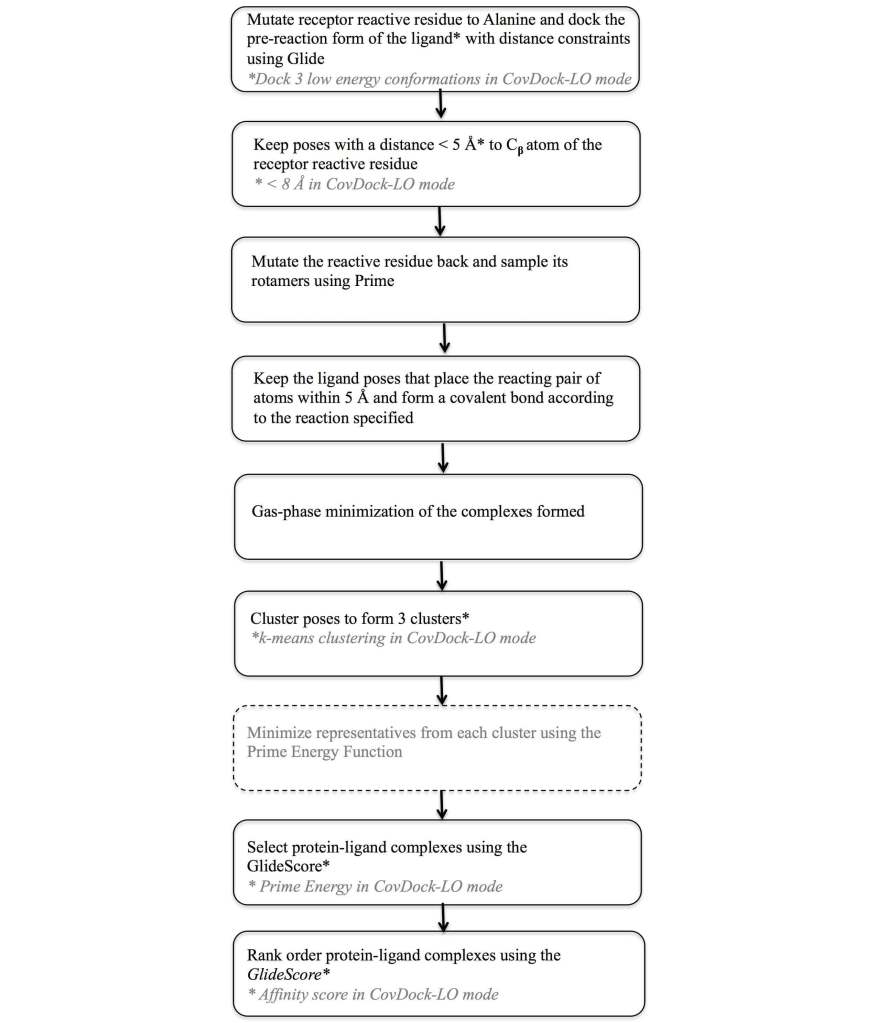
To address this need a covalent docking protocol, CovDock13, 14 was developed that yields good accuracy in binding mode prediction and has automatic detection of reactive atoms using SMARTS patterns. Using a workflow that combines Glide docking and Prime optimization, the CovDock-VS (virtual screening) and CovDock-LO (lead optimization) modes enable application of covalent docking in large-scale virtual screening or lead optimization projects, respectively. The workflow for these two approaches is shown in Figure 7. Thanks largely to reduced sampling, the CovDock-VS mode is 10 to 40 times faster then CovDock-LO mode.
Docking accuracy for CovDock is good with an average RMSD of 1.9Å for CovDock-VS and 1.5 Å for CovDock-LO in a retrospective native docking experiment with results shown in Table 4. In combination with interaction filters (in these cases, H-bond constraints), CovDock-VS has also demonstrated excellent enrichment in retrospective experiments as shown in Table 5. In a recent test it has been shown to retrieve 71%, 72%, and 77% of the known actives for Cathepsin K, HCV NS3 protease, and EGFR within 5% of a decoy library, respectively. Even in cases where interaction filters could not be used, as for XPO1, CovDock-VS was still able to retrieve 95% of known actives within the top 30% of decoy compounds, with 33% of known compounds found in the top 1% of decoys.
| CovDock-VS | CovDock-LO | ||
|---|---|---|---|
| Cathepsin K actives | |||
| 1YT7-ligandc | 2.68 | 0.68 | |
| 2BDL-ligand | 2.48 | 2.89a | |
| 1TU6-ligand | 1.43 | 3.18 | |
| HCV NS3 protease actives | |||
| 3KNX-ligand | 2.89a | 2.45 | |
| 2GVF-ligand | 1.66 | 1.51 | |
| 3LON-ligand | 6.69a | 1.81 | |
| 2OC7-ligand | 1.38 | 1.12 | |
| 2F9U-ligand# | 1.19 | 1.30 | |
| 2OBO-ligand | 1.29 | 0.98 | |
| 2OC8-ligand | 1.55 | 1.04 | |
| 2A4Q-ligand | 2.51 | 2.28 | |
| 2OC0-ligand | 3.03 | 0.88 | |
| 3LOX-ligand | 2.07 | 1.72 | |
| 2OC1-ligand | 3.75a | 3.14 | |
| EGFR actives | |||
| 4I24-ligand (Dacomitinib) | 1.46 | 0.85 | |
| 4G5J-ligand (Afatinib) | 1.81 | 0.86 | |
| 3IKA-ligand (WZ4002) | 2.53a | 1.67 | |
| 2JIV-ligand (Neratinib) | 6.54a | 2.41a | |
| 2J5F-ligand (34-JAB) | 1.57 | 1.20 | |
| XPO1 actives | |||
| 4GMX-ligand (KPT-185)c | 2.19b | 0.67 | |
| KPT-276 (not submitted to PDB)c | 2.54a | 1.07 | |
| Mean | 1.87 | 1.50 | |
| EF1% | EF10% | BEDROC (α = 20) | |
|---|---|---|---|
| HCV NS3 protease | 52 | 7 | 0.70 |
| Cathepsin K | 9 | 8 | 0.48 |
| EGFR | 46 | 8 | 0.65 |
| XPO1 | 33 | 7 | 0.52 |
References
-
Friesner, R. A.; Banks, J. L.; Murphy, R. B.; Halgren, T. A.; Klicic, J. J.; Mainz, D. T.; Repasky, M. P.; Knoll, E. H.; Shelley, M.; Perry, J. K.; Shaw, D. E.; Francis, P.; Shenkin, P. S., “Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy”,
J. Med. Chem., 47, 1739-49 (2004).
-
Halgren, T. A.; Murphy, R. B.; Friesner, R. A.; Beard, H. S.; Frye, L. L.; Pollard, W. T.; Banks, J. L., “Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening”,
J. Med. Chem., 47, 1750-59 (2004).
-
Friesner, R. A.; Murphy, R. B.; Repasky, M. P.; Frye, L. L.; Greenwood, J. R.; Halgren, T. A.; Sanschagrin, P. C.; Mainz, D. T., ”Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes”,
J. Med. Chem., 49, 6177-96 (2006).
-
Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J. Y.; Wang, L.; Lupyan, D.; Dahlgren, M. K.; Knight, J. L.; Kaus, J. W.; Cerutti, D. S.; Krilov, G.; Jorgensen, W. L.; Abel, R.; Friesner, R. A., ”OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins”,
J. Chem. Theory Comput., 12(1), 281-296 (2016).
-
Hartshorn, M. J.; Verkonk, M. L.; Chessari, G.; Brewerton, S. C.; Mooij, W. T. M.; Mortenson, P. N.; Murray, C. W., ”Diverse, High-Quality Test Set for the Validation of Protein-Ligand Docking Performance”,
J. Med. Chem., 50, 726-741 (2007).
-
Huang, N.; Shoichet, B. K.; Irwin, J. J., ”Benchmarking Sets for Molecular Docking”,
J. Med. Chem., 49, 6789-6801 (2006).
-
Repasky, M. P.; Murphy, R. B.; Banks, J. L.; Greenwood, J. R.; Tubert-Brohman, I.; Bhat, S.; Friesner, R. A., ”Docking performance of the Glide program as evaluated on the Astex and DUD datasets: a complete set of Glide SP results and selected results for a new scoring function integrating WaterMap and Glide”,
J. Comput. Aided Mol. Des., 26, 787-99 (2012).
-
Mysinger, M. M.; Carchia, M.; Irwin, J. J.; Shoichet, B. K., ”Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking”
J. Med. Chem., 55, 6582-94 (2012).
-
Kuhn, B.; Guba, W.; Hert, J.; Banner, D.; Bissantz, C.; Ceccarelli, S.; Haap, W.; Korner, M.; Kuglstatter, A.; Lerner, C.; Mattei, P.; Neidhart, W.; Pinard, E.; Rudolph, M. G.; Schulz-Gasch, T.; Woltering, T.; Stahl, M., ”A Real-World Perspective on Molecular Design”
J. Med. Chem., 9(9), 4087–4102 (2016).
-
Tubert-Brohman, I.; Sherman, W.; Repasky, M.; Beuming, T., ”Improved docking of polypeptides with Glide”,
J. Chem. Inf. Model., 53, 1689-99 (2013).
-
Feher, M.; Williams, C. I., ”Numerical errors and chaotic behavior in docking simulations”,
J. Chem. Inf. Model., 52(3), 724–738 (2012).
-
Robertson, J. G., ”Mechanistic basis of enzyme-targeted drugs”,
Biochemistry, 44, 5561-71 (2015).
-
Toledo Warshaviak, D.; Golan, G.; Borrelli, K. W.; Zhu, K.; Kalid, O., ”Structure-based virtual screening approach for discovery of covalently bound ligands”,
J. Chem. Inf. Model., 54, 1941-50 (2014).
-
Toledo Warshaviak, D.; Golan, G.; Borrelli, K. W.; Zhu, K.; Kalid, O., ”Docking covalent inhibitors: a parameter free approach to pose prediction and scoring”,
J. Chem. Inf. Model., 54, 1932-40 (2014).
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.