Schrödinger solutions for small molecule protonation state enumeration and pKa prediction
Executive Summary
The pKa of a drug is a key physicochemical property to consider in the drug discovery process given its importance in determining the ionization state of a molecule at physiological pH. Schrödinger provides several solutions for predicting pKa values, protonation state distribution and derived properties that can be applied across a range of drug discovery stages, from screening through lead optimization. Here we provide an overview of each technology solution and use case examples of how they can be applied in drug discovery.
Background
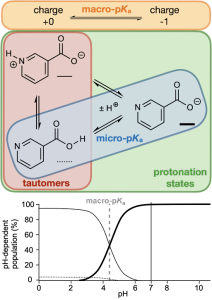
Small molecules can undergo ionization in solution where they either lose or gain protons (H+) at different ionizing sites. The measure of the propensity of a site or molecule to ionize by the association/dissociation of one or more protons is quantified by a pKa value. If the pKa value refers to a particular site ionizable site the value is a microscopic pKa (micro-pKa), and it is a macroscopic pKa (macro-pKa) if the value refers to the entire molecule. The specific arrangement of protons around the ionizing sites constitutes a protonation state, and different protonation states of the same charge level are called tautomers. Each protonation state is in thermodynamic equilibrium with the others and therefore has a free energy associated with its population within this collection of protonation states, which may be derived either from micro-pKa values through thermodynamic equations or obtained directly by comparing the free energies of the states. In drug design, understanding the different protonation states of a molecule is critical, since they will drive properties including solubility, membrane permeability, and activity.
Challenges of pKa Prediction
Determining which states predominate at a given pH and by how much is a challenging task both experimentally and computationally because the number of states that are all in thermodynamic equilibrium grows ~2n with the number, n, of singly protonatable sites. Thus, molecules with many titratable sites can potentially have a large number of different protonation states, all of which need to be enumerated and energetically scored.
Computationally, Schrödinger uses two main approaches to score states: 1) through evaluating thermodynamic equilibrium equations with micro-pKa values, and 2) directly predicting the states’ relative free energies. Predicting pKa values is an important step to calculating state distributions, which in turn enables prediction of important related quantities that would otherwise be inaccessible.
Overview of Schrödinger Solutions
Epik Classic
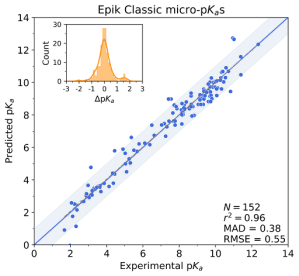
Epik Classic, previously known simply as Epik1, is an expert system for rapidly and accurately predicting the micro-pKa values and the most populated protonation states for a ligand at a given pH. The underlying pKa prediction technology is the empirical Hammett-Taft linear free energy relationship (LFER), which identifies an ionizing group, takes its root pKa value, perturbs it by the bonded chemical fragments, and applies charge spreading to arrive at its effective micro-pKa value. Epik Classic then uses the predicted pKa values to enumerate a ligand’s protonation states, rank them by energy, and then return the most populated states. Because Epik Classic uses SMARTS patterns-based rules, it is fast enough for high-throughput, although at the expense of being unaware of both conformational and stereochemical effects.
Epik 7
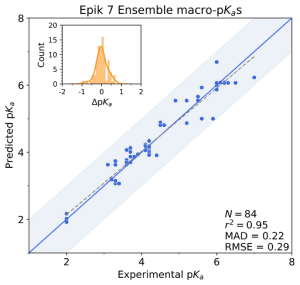
Epik 7 is a complete redesign of Epik that leverages Schrödinger’s powerful machine learning (ML) technology for more accurate results across broader chemical space. Ionizing groups are initially identified by SMARTS patterns and are then used to enumerate the protonation states for a range of ionizations.2 The micro-pKa values of each site in each state are predicted with 3-layer atomic graph convolutional neural networks (GCNNs) extending out radially six bonds from the ionizing atom. The predicted pKa values for the states are then used to predict the relative energies of the states to both allow determination of the most populated states at a pH and calculation of macro-pKa values. The topological nature of the ML approach means that Epik 7, like previous versions, is rapid but agnostic to 3D geometry and stereochemistry.
Jaguar pKa
Jaguar pKa takes a third, more physics-based approach to predicting micro-pKa values for a ligand. This workflow calculates the pKa values at the user-defined ionizing sites in a query ligand by first generating the conjugate pair, on which are then executed conformational searches to locate the lowest energy structures,3 followed by density functional theory (DFT) based geometry optimizations and single-point energy evaluations. These resulting conformationally-averaged, “raw” micro-pKa values are then corrected using empirically-parametrized relationships to give accurate predictions. Jaguar pKa performs best on non-tautomerizable structures. Being physics-based, it does take into account geometric and stereochemical effects, but at the expense of speed.
Macro-pKa
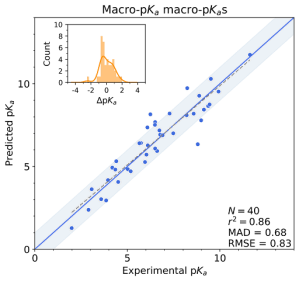
Macro-pKa follows the same philosophy as Jaguar pKa by combining physics-based DFT calculations with empirical corrections, but extends its applicability to enable calculation of tautomerizable ligands. Macro-pKa automatically identifies ionizing sites, enumerates the protonation states, and calculates the micro-pKa values following a similar workflow to Jaguar pKa, but with an enhanced scheme for generating empirical corrections. Finally, the calculated micro-pKa values are used to rank the protonation states by energy, return the most populated states for a user-supplied pH, and determine the macro-pKa values for the ligand. The exhaustiveness of this approach comes at a larger time and resource cost than Jaguar pKa.
Use Cases
Here we outline several use cases for pKa prediction in the drug discovery workflow.
Note: Each use case example outlines below could be approached with any of the listed solutions within that section. The dataset presented highlights the applicability of just one of the possible solutions.
I. Querying microscopic pKa values
Applicable Solutions
- Epik Classic
- Epik 7
- Jaguar pKa
When investigating the binding modes of a ligand, the micro-pKa value of an ionizing site is an indicator of the propensity for it to become ionized at a given pH. The ionization state of the ligand directly influences how it interacts with another molecule such as a protein, e.g., whether or not it can participate in a salt bridge.

II. Querying apparent or macroscopic pKa values
Applicable Solutions
- Epik 7
- Macro-pKa
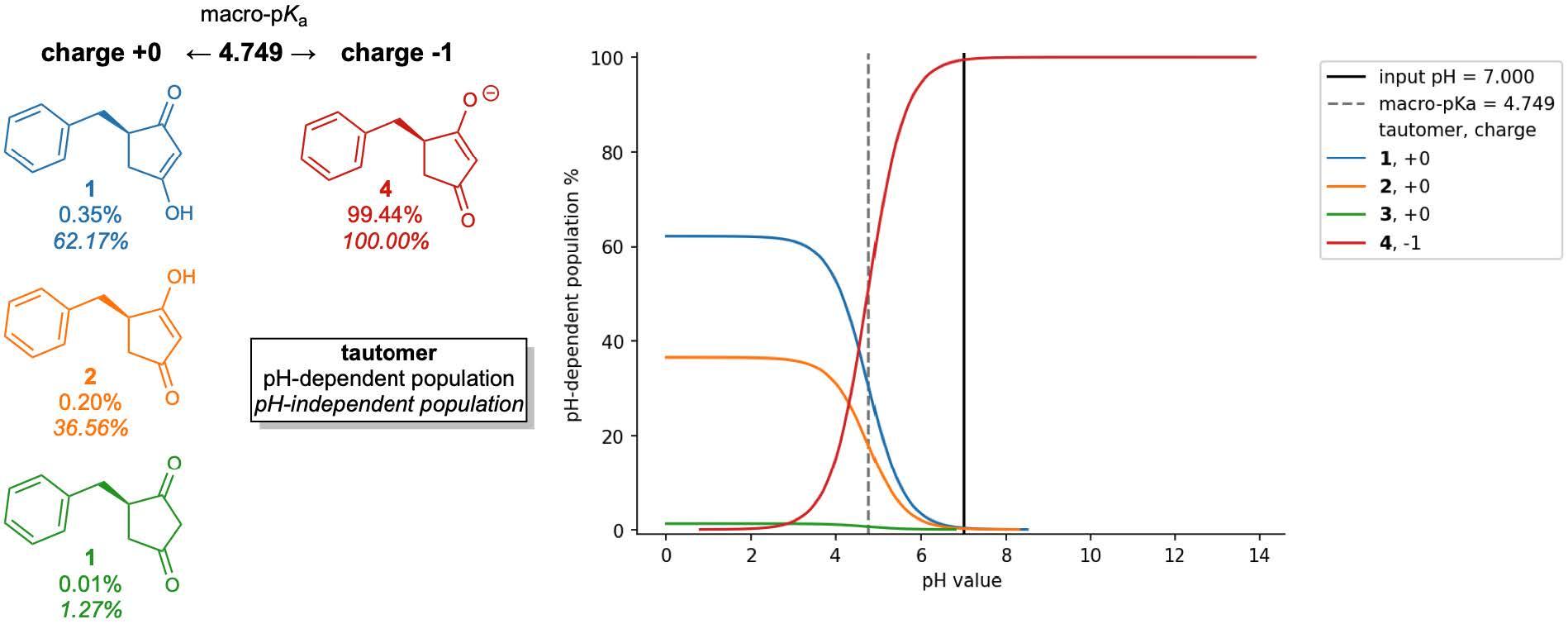
For monoprotic or polyprotic compounds with a single dominant tautomer at each charge level, micro-pKas may very closely match the apparent or macro-pKa value that is most commonly obtained through titration experiments. However, for compounds or ionization states with multiple competitive tautomers, the micro-pKa value of a single tautomer may not fully reproduce the experimentally observed macroscopic value. To obtain this apparent value, all states’ must first be enumerated and evaluated so that all their micro-pKa values are considered in the macro-pKa calculation.
III. Ligand preparation and high-throughput screening
Applicable Solutions
- Epik Classic
- Epik 7
Physics-based simulations typically require specification of all atoms in the simulation system, including all hydrogen atoms. Thus, structure-based simulations including Glide docking, molecular dynamics, and free energy perturbation with FEP+ should be performed using an ensemble of the highly-populated protonation states of a ligand. Therefore, a crucial first step in any structure-based screen of a small molecule ligand library is to prepare the ligands by obtaining the most populated protonated states. Epik Classic and Epik 7 are integrated with our automated ligand preparation workflow, LigPrep, to allow preparation of large ligand libraries for high-throughput screening. Additionally, both Epik Classic and Epik 7 and their LigPrep implementations allow for the generation and scoring of additional states that may potentially bind to metal ions in the pocket.
IV. Hit-to-lead optimization
Applicable Solutions
- Epik Classic
- Epik 7
Once hits are identified, a series of analogs are synthesized to explore the relevant chemical space in greater detail to arrive at improved behavior. It is important to be able to screen potential candidates rapidly and accurately to assess which to optimize further. The < 0.5 log unit accuracy and sub-second calculation speed of Epik Classic and Epik 7 make them excellent tools for rapid idea generation and testing. In addition to pKa value and protonation state distribution prediction, they have been implemented in other ADMET or property predictors, such as for membrane permeability and solvation energy.
V. Early-stage lead optimization
Applicable Solutions
- Epik 7
- Jaguar pKa
- Macro-pKa
Optimizing the many physical characteristics required can be laborious and costly, from ideation, through synthesis and assay. In this environment, where high quality property predictions are required and time permits, Schrödinger’s physics-based predictors, Jaguar pKa and Macro-pKa, take into account more molecular characteristics, including conformational and stereochemical effects to improve pKa prediction accuracy. Additionally, Macro-pKa and Epik 7 both offer detailed speciation reports for a queried ligand. These are especially helpful for understanding the distribution of tautomeric states across the pH spectrum.
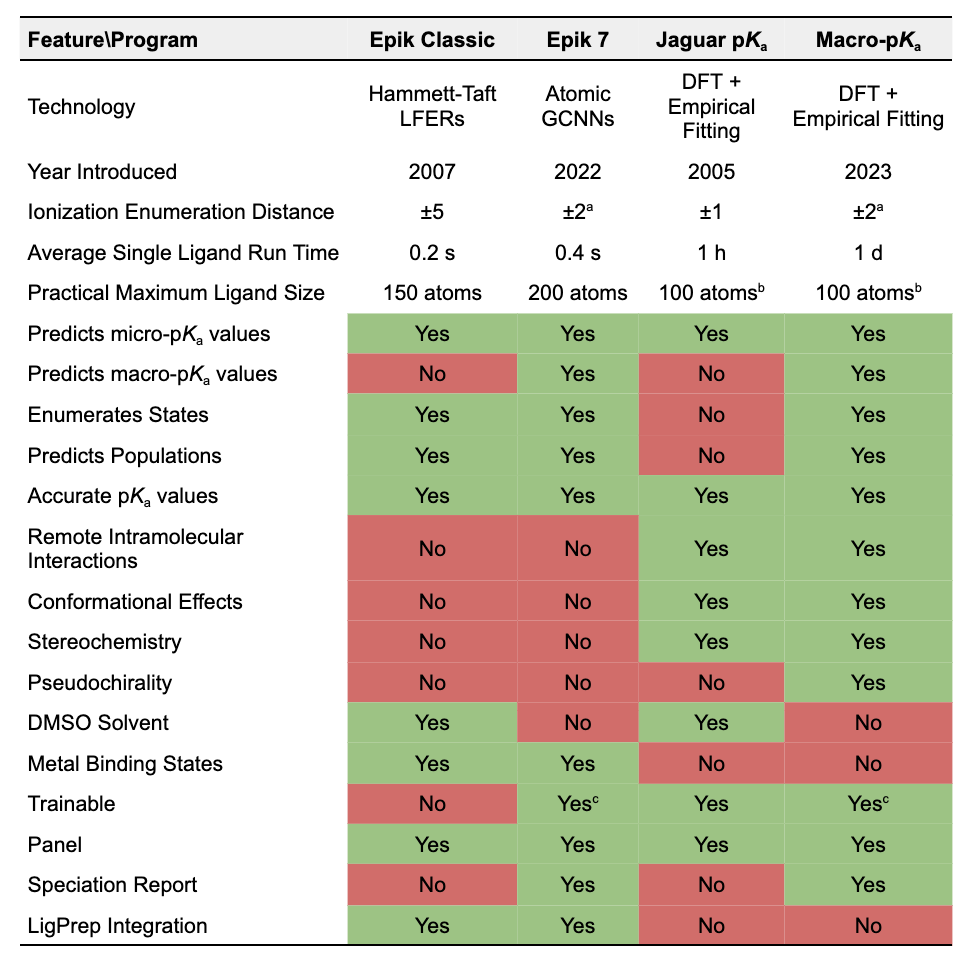
Feature Comparison Table
References
-
Epik: A Software Program for pKa Prediction and Protonation State Generation for Drug-like Molecules.
Shelley, J. C. et al. J. Comput. Aided Mol. Des. 2007, 21 (12), 681–691
-
Epik: pKa and Protonation State Prediction through Machine Learning.
J. Chem. Theory Comput. 2023, 19 (8), 2380–2388
-
Multiconformation, Density Functional Theory-Based pKa Prediction in Application to Large, Flexible Organic Molecules with Diverse Functional Groups.
Bochevarov, A. D. J. Chem. Theory Comput. 2016, 12 (12), 6001–6019.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.