Force Field Bundle
Improve the quality of your computational predictions with best-in-class, proprietary force fields — developed in-house and built for accuracy

Improve the quality of your computational predictions with best-in-class, proprietary force fields — developed in-house and built for accuracy
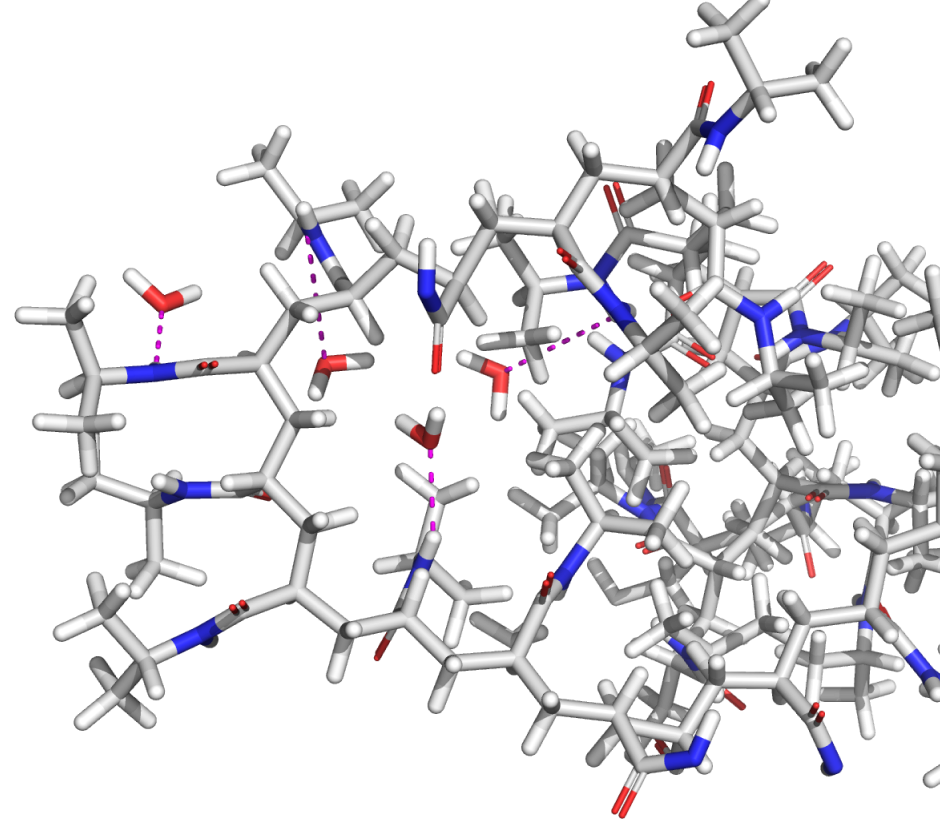

Force fields are used in molecular simulations to describe the interactions between atoms in a system. Having an accurate force field is at the heart of obtaining useful molecular structures and predicting relative energies, and yet many in silico programs employ force fields that are years, if not decades, old and suffer from lack of sufficient coverage for many common molecular motifs.

We are witnessing a shift toward a computational “predict-first” approach to drug discovery and materials science research.

Generate precise binding free energy predictions with FEP+, enabling more reliable rank ordering within congeneric series.
Enhance and accelerate physics-based computational methods by integrating AI/ML into force fields and simulation engines.
Accurately predict binding modes of novel scaffolds using advanced induced fit docking methods in IFD-MD.
Reveal mechanisms of action and key interaction energies through high-fidelity molecular dynamics simulations with Desmond.
Achieve better conformational sampling and docking poses with improved torsional energy descriptions across Glide, ConfGen, MacroModel, and Prime.

Accurate ligand binding mode prediction for novel chemical matter, for on-targets and off-targets
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
Efficient tool for optimizing custom torsion parameters in OPLS4
Cutting-edge force field technologies for accurate property predictions
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.