Desmond
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy

High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
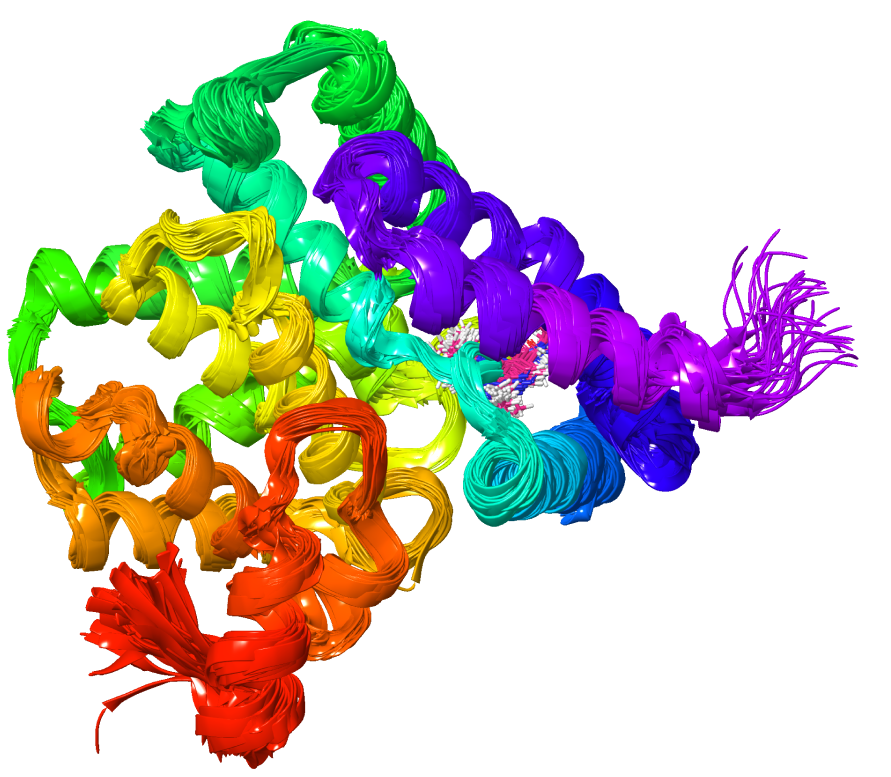
Desmond is a GPU-powered high-performance molecular dynamics (MD) engine for simulating biological systems such as small protein, viral capsids, protein-ligand complexes, small molecules in mixed solvents, organic solids, and synthetic macromolecular complexes.
Achieves exceptional throughput on commodity Linux clusters with both typical and high-end networks and improves computing speed by 100x on general-purpose GPU (GPGPU) compared to single CPU
Constructed with a focus on numerical accuracy, stability, and rigor, Desmond’s performance enables the simulation of large-scale features of nanometer to micron size over time scales of picoseconds to microseconds
Provides a robust framework for the calculation of energies and forces for atomistic force field models and is compatible with chemistries commonly used in biomolecular research
Performs explicit solvent simulations with periodic boundary conditions using simulation boxes with careful attention to the calculation of long-range electrostatics, and can be used to model protein and nucleic acid systems with explicit lipid membranes
Provides intelligent default settings and allows for rapid setup of computational experiments in an intuitive interface, while supporting automated simulation setup including system building, analysis tools, and force field assignment
Enables visualization and examination of computed results within the same Maestro modeling environment that connects to a comprehensive suite of modeling tools from quantum mechanics to machine learning
Improved cryptic pocket identification through enhanced sampling. Leverage MxMD with our new interface for simplified setup, analysis, and customizable visualization of cryptic binding pockets on protein surfaces.

Characterize ligand-receptor interactions with unbinding kinetics analysis. Visualize unbinding pathways using enhanced sampling methods to identify and optimize promising lead compounds based on their dissociation rates.
Schrödinger has a strategic partnership with NVIDIA to optimize our computational drug discovery platform for NVIDIA GPU technology.
Get answers to common questions and learn best practices for using Schrödinger’s software.

Learn more about the related computational technologies available to progress your research projects.
Secure, scalable environment for running simulations on the cloud
A modern, comprehensive force field for accurate molecular simulations
Accurate ligand binding mode prediction for novel chemical matter, for on-targets and off-targets
Discover how Schrödinger technology is being used to solve real-world research challenges.
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.