High precision, computationally-guided discovery of highly selective Wee1 inhibitors for the treatment of solid tumors
Design approach leverages rigorous physics-based modeling to overcome off-target liabilities en route to development candidate SGR-3515.
in silico
Performed rapid in silico design cycles using a collaborative platform and a large-scale de novo design workflow
FEP+
Optimized potency and selectivity with relative binding FEP+ and protein FEP+
Candidate
Resulted in a development candidate currently in preclinical development
Wee1, Ser/Thr kinase
Schrödinger proprietary program, small molecule
Solid tumors
IND-enabling studies
“This program demonstrates the first prospective application of protein FEP+ to model broad kinome selectivity, which not only enabled the Wee1 team to rapidly identify a few highly selective chemotypes and rescue a program that had hit a major roadblock, but it also added a powerful tool for the broader drug discovery community to tackle selectivity challenges more efficiently.”
Jiashi Wang
Senior Director, Medicinal Chemistry
Schrödinger Therapeutics Group
Design challenge
Inhibition of Wee1, a serine/threonine protein kinase which serves as the gatekeeper of the G2-M cell-cycle checkpoint, forces cells into unscheduled mitosis and culminates in cell death. Given its critical role in DNA repair, Wee1 is an attractive target for oncology drug development. Clinical trial data for the Wee1 inhibitor adavosertib (AZD-1775) validated the potential of targeting Wee1 by revealing strong anti-cancer activity in solid tumors.1-11 However, it was suspected that AZD-1775 had potential liabilities due to inhibition of several other kinases, including the PLK family, as well as time-dependent inhibition of CYP3A4.
The aim of this program, driven by Schrödinger Therapeutics Group, was to develop a best-in-class, highly selective Wee1 inhibitor by leveraging rigorous physics-based modeling approaches to address off-target liabilities with high precision.
Efficient generation of a selective and potent lead series with a rigorous FEP-based workflow
At project onset, the team’s goal was to identify one or multiple novel lead series with improved selectivity for Wee1 as compared to polo-like kinase 1 (PLK1). The team enumerated over six million ideas and hand-drawn designs, then triaged them using an automated chemistry-based filtering workflow, structure-based screening (docking), and ultimately rigorous calculation of relative binding free energies via free energy perturbations (FEP+, De Novo Design Workflow) (Figure 1). The accuracy and utility of FEP+ as a computational binding affinity assay has been validated extensively, generating predictions within 1.0 kcal/mol of experimental values on average.12 By combining FEP+ with high performance cloud computing and machine learning (Active Learning FEP+), over 9,000 FEP+ calculations were performed to evaluate the affinity of the most promising designs for inhibiting Wee1 and ensuring selectivity over PLK1.
Design ideas were then evaluated by the project team in LiveDesign, Schrödinger’s cloud-based enterprise informatics platform. These workflows informed the selection of less than 100 compounds for synthesis and testing, resulting in the identification of multiple novel chemotypes with nanomolar affinity and enhanced selectivity (up to 1000x) for Wee1 over PLK1 within seven months from project start.13
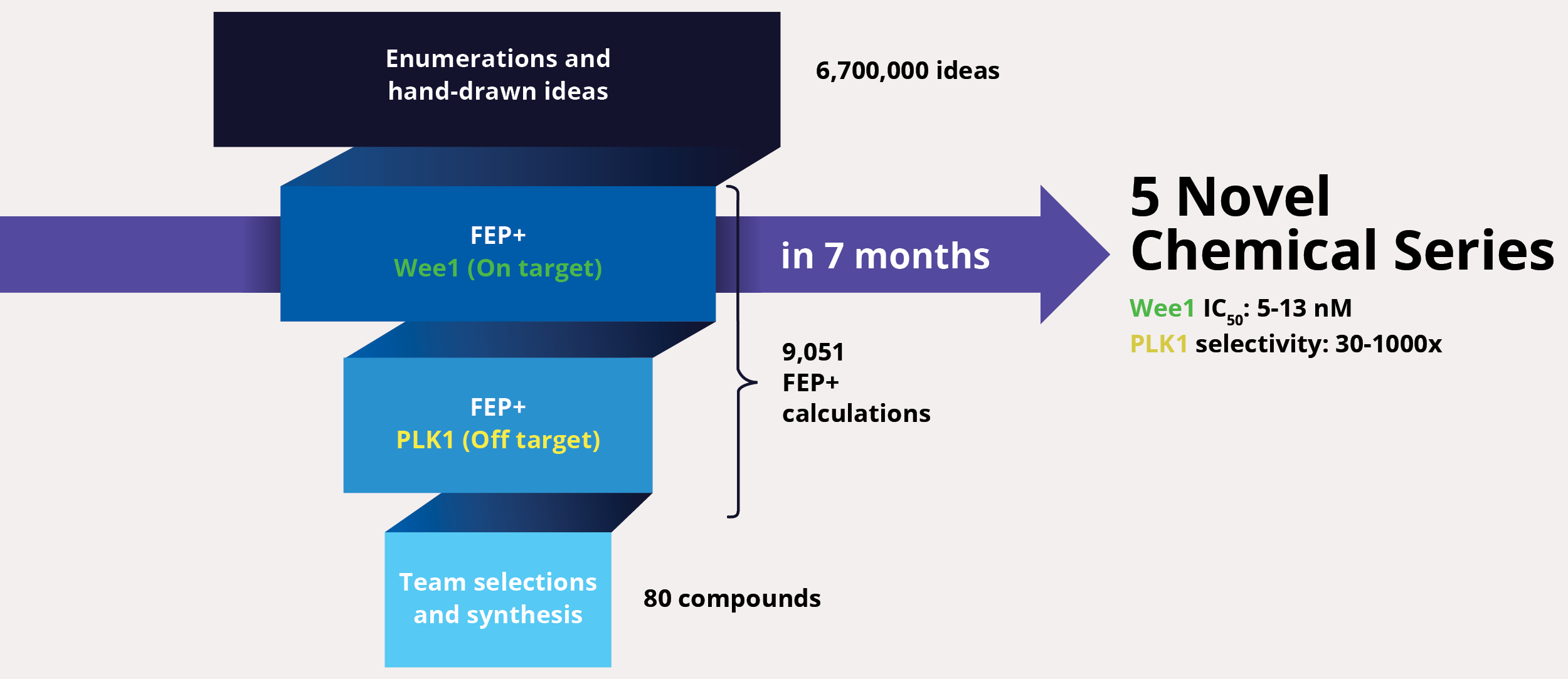
Addressing additional off-target liabilities with protein FEP+
Although the team successfully improved selectivity against PLK1 in the first phase of the project, subsequent kinase panel screening revealed a significant number of unanticipated off-target kinase liabilities. In lieu of starting over or scrapping the program entirely, the team explored a complementary strategy to improve selectivity of the chemotypes identified in the first phase. A major driver of selectivity seemed to be a specific residue in the binding site. The team utilized protein FEP+ calculations, a protocol within FEP+, to assess the impact of single point-mutations at that location on the ligand affinity and to infer selectivity across a large diversity of kinases without the need to profile each kinase separately. In this new workflow, 6,700 new designs were profiled with ligand FEP+ for predicting potency against Wee1 and with protein FEP+ for predicting broad kinome selectivity. During this three month modeling campaign, the ligand FEP+ and protein FEP+ strategy identified 42 promising molecules for synthesis and 22 of these molecules exhibited low nanomolar to picomolar measured potencies against Wee1 with substantially reduced selectivity liabilities (Figure 2).14
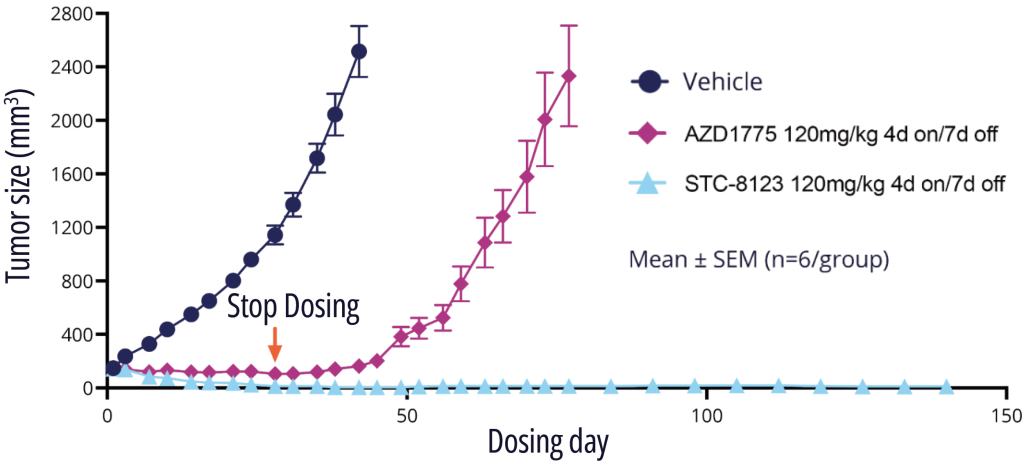
One exquisitely selective molecule, STC-8123, was used as a proof-of-concept compound to demonstrate that highly selective Wee1 inhibitors retain profound in vivo efficacy. Indeed, in an A427 mouse model with intermittent dosing, STC-8123 demonstrated rapid and more sustainable inhibition of tumor growth compared with AZD-1775 (Figure 3).14

Optimizing DMPK and ADME properties with machine learning and quantum mechanics strategies
With a promising chemical series in hand, the team began the process of optimizing DMPK and ADME properties. As experimental data accumulated for the lead series, the team trained project-specific machine learning (ML) models to predict time-dependent inhibition of CYP3A4 profiles as well as a variety of ADME properties (DeepAutoQSAR). In addition, quantum mechanical (QM) calculations were developed for modeling compound reactivity with the CYP3A4 heme, a potential target for drug-drug interaction liabilities (Jaguar). During late-stage lead optimization, this breadth of ML models and QM physics-based calculations enabled prospective multiparameter optimization to readily narrow down the chemical space that was sent for synthesis and testing.
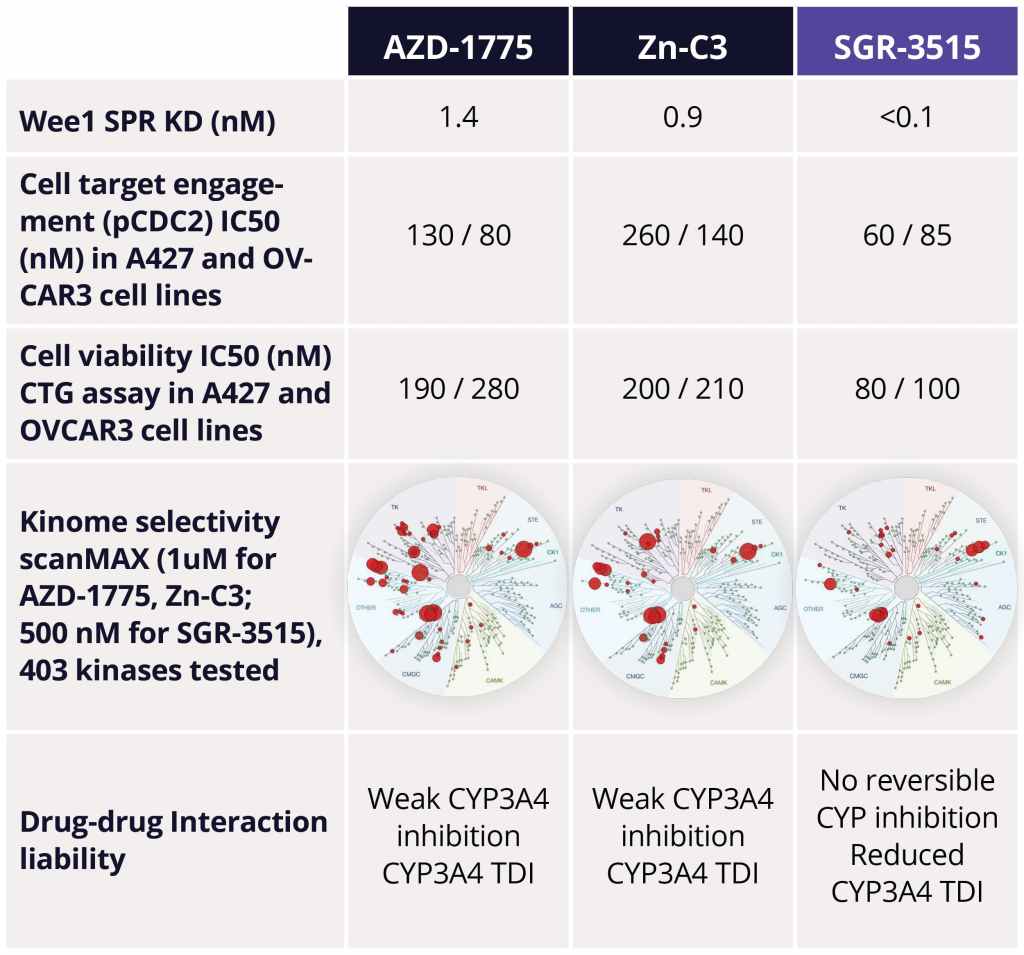
The team identified several advanced leads from among the ~300 compounds synthesized in the series. Upon in-depth profiling of advanced leads and careful consideration of the properties that a best-in-class, next generation Wee1 inhibitor should have, SGR-3515 was nominated as the development candidate. SGR-3515 is an exquisitely selective, potent molecule that is structurally-differentiated from competitors’ molecules, has a differentiated ADME profile, and achieves superior in vivo efficacy (Figure 4).
Enabling digital technologies to drive discovery programs
FEP+
Elucidation of ligand binding preferences for large families of off-targets with protein FEP+ and on-target potency with FEP+
De Novo Design Workflow
Ultra-large scale chemical space exploration combining multiple compound enumeration strategies with an advanced filtering cascade.
DeepAutoQSAR
Scalable, intuitive tool for training and running ML/AI models automatically using molecular inputs
LiveDesign
Collaborative enterprise informatics platform for centralizing access to virtual and wet lab project data and powerful computational predictions.
References
-
Clin Cancer Res 2021: 27(4).
-
J Clin. Oncol 2021: 39(14):1531-1539.
-
J Clin. Oncol 40, no. 16_suppl (June 01, 2022) 5515.
Yin et al. ASH Presentation 2021.
-
Lancet 2021: 398:281-92.
-
J Clin. Oncol 2016: 34:4354-4361.
-
Clin Cancer Res 2018: 24(120): 2740-8.
-
Clin Cancer Res 2018: 15;24(12):2740-2748.
-
Zentalis investor deck.
-
Annals of Oncology 2020: 31 (suppl_4).
-
J Clin Oncol 2019 Oct 10;37(29).
-
Cancer Res (2019) 79 (13_Supplement): CT02. 12.J Clin Oncol 2021 Nov 20;39(33).
-
Advancing drug discovery through enhanced free energy calculations. Abel et al.
Acc. Chem. Res. 2017, 50, 7, 1625–1632.
-
De-risking off-target liabilities with protein free energy methods.
Knight et al. ACS 2022.
-
Discovery of potent, selective, and orally available WEE1 inhibitors that demonstrate increased DNA damage and mitosis in tumor cells leading to tumor regression in vivo.
Sun et al. AACR 2022.
-
Into the clinic: Transforming the drug discovery process with digital chemistry.
Davis et al. Lab of the Future 2023.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.