FEP+
High-performance free energy calculations for drug discovery

High-performance free energy calculations for drug discovery
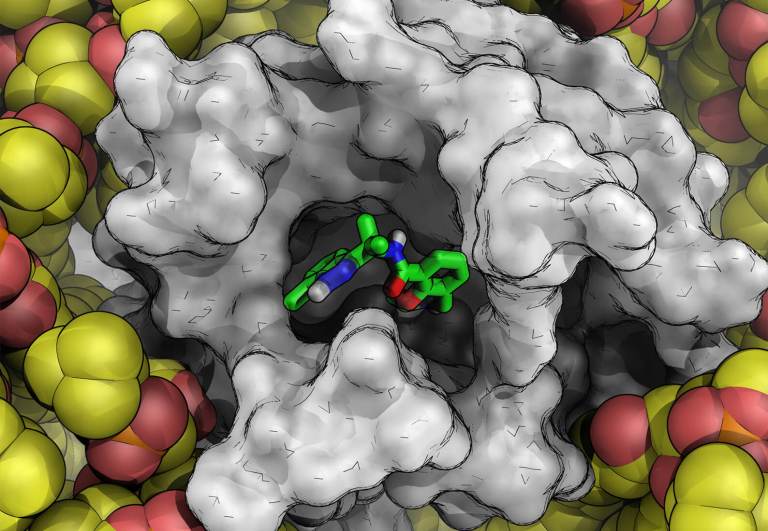
FEP+ is Schrödinger’s proprietary, physics-based free energy perturbation technology for computationally predicting protein-ligand binding at an accuracy matching experimental methods, across broad chemical space.
Leverage FEP+ as an accurate in silico binding affinity assay to drive rapid virtual design cycles and focus experimental efforts on only the highest quality ideas

Optimize multiple properties simultaneously, including potency, selectivity, and solubility, to improve the profile and developability of small and large molecules

Synthesize novel and challenging chemistry with a high degree of confidence through prospective application of FEP+

Predictive accuracy approaching experiment (1 kcal/mol) as demonstrated in large-scale validation studies across diverse ligands and protein classes
Widely adopted by leading pharma and biotech companies, with several drug candidates in the clinic driven by FEP+
Supports the broadest range of applications and perturbation types common in drug discovery scenarios and consistently expanded through active R&D
 Validate protein models without experimental structures or from low resolution structures using IFD-MD with FEP+ Structurally enable off-targets and design out common ADMET liabilities
Validate protein models without experimental structures or from low resolution structures using IFD-MD with FEP+ Structurally enable off-targets and design out common ADMET liabilities  Rescore hits from virtual screens to prioritize synthesis lists and improve using absolute binding FEP+
Leverage available chemical matter to efficiently discover novel cores via core hopping
Perform large-scale in silico fragment screens using absolute binding FEP+ and solubility FEP+
Rescore hits from virtual screens to prioritize synthesis lists and improve using absolute binding FEP+
Leverage available chemical matter to efficiently discover novel cores via core hopping
Perform large-scale in silico fragment screens using absolute binding FEP+ and solubility FEP+ 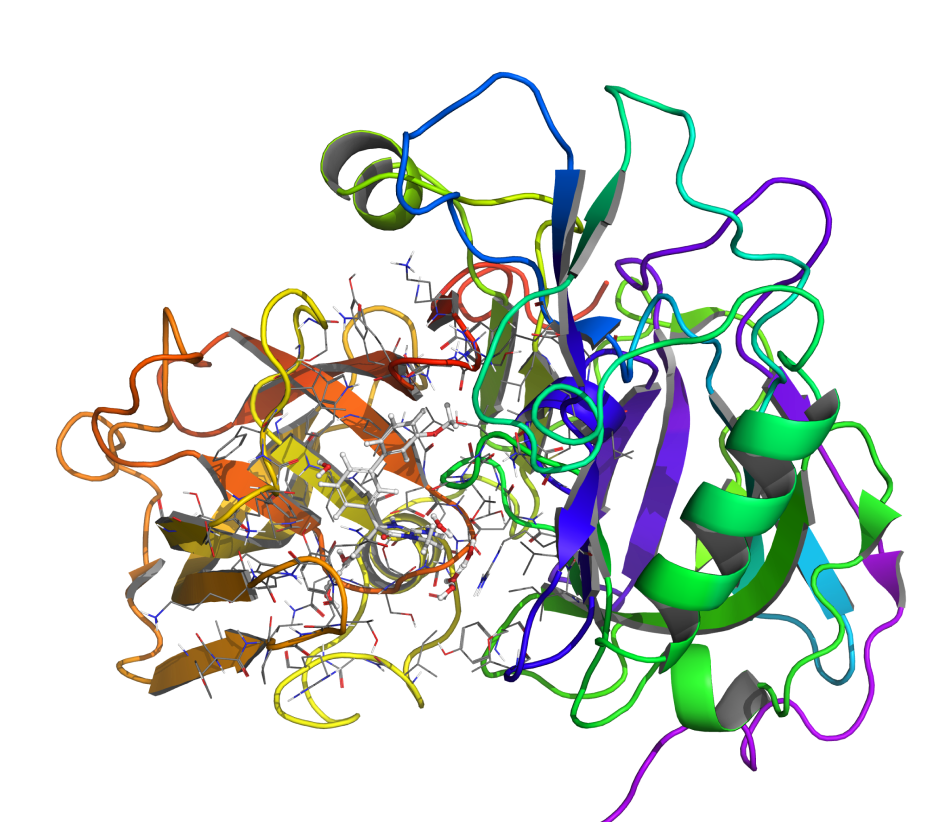 Rapidly optimize on-target potency by leveraging FEP+ as an in silico binding affinity assay
Optimize selectivity to known off-targets and across large gene families
Maintain on-target potency and selectivity while optimizing ADMET properties
Rapidly optimize on-target potency by leveraging FEP+ as an in silico binding affinity assay
Optimize selectivity to known off-targets and across large gene families
Maintain on-target potency and selectivity while optimizing ADMET properties 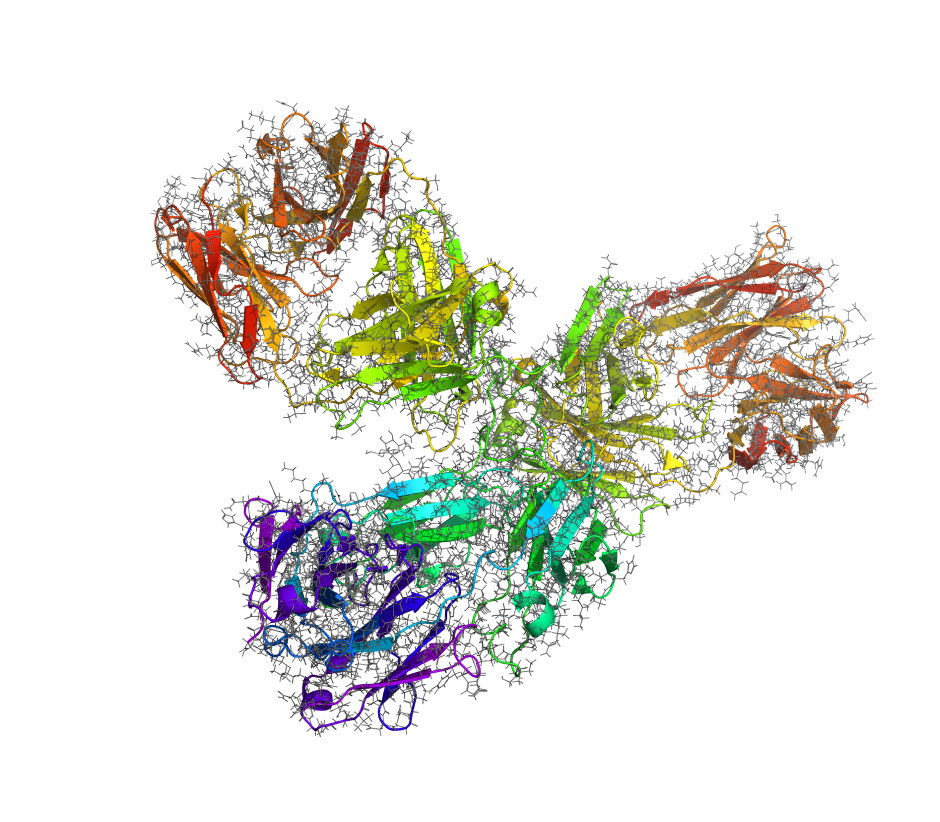 Refine antibody candidate selection with accuracy that reproduces experimentally determined relative free energies Predict binding affinity, selectivity, and thermostability of peptides Engineer enzymes for substrate selectivity and specificity
Refine antibody candidate selection with accuracy that reproduces experimentally determined relative free energies Predict binding affinity, selectivity, and thermostability of peptides Engineer enzymes for substrate selectivity and specificity 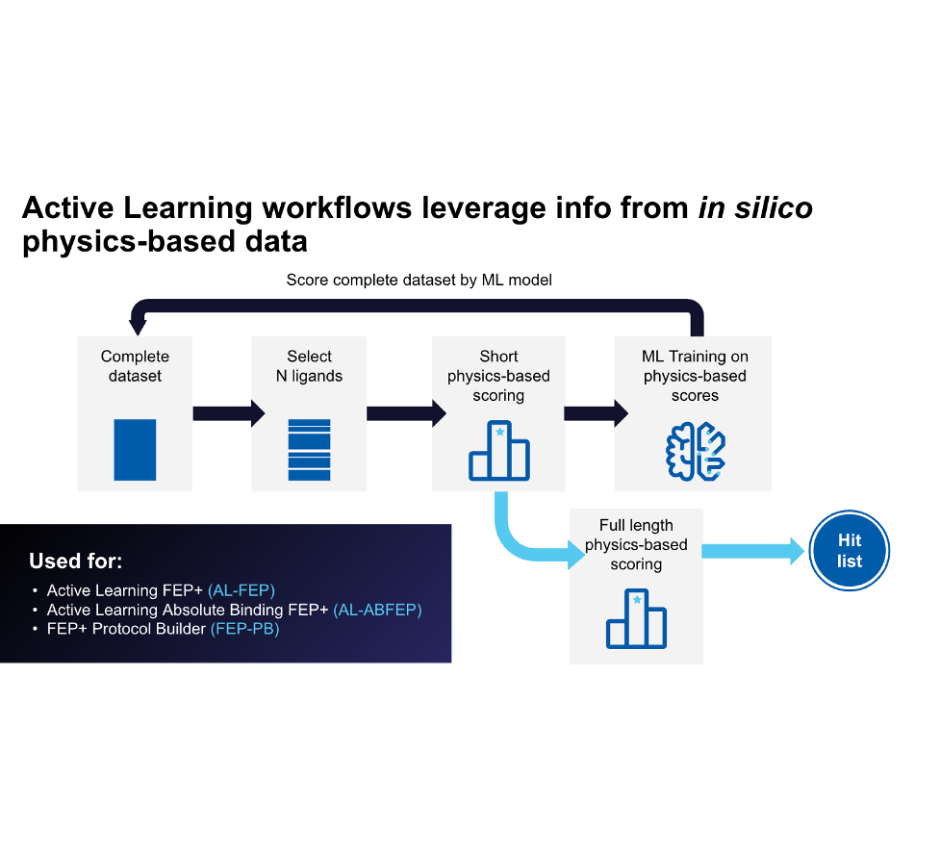
Leverage a well-validated, automated workflow which trains a machine learning model on project-specific FEP+ data to allow processing of up to millions of compounds with highly accurate FEP+ calculations efficiently.
 Blog
Life Science
Blog
Life Science
Level-up your FEP+ skills and enroll in our online molecular modeling course, Free Energy Calculations for Drug Design with FEP+.
View CourseDiscover how Schrödinger technology is being used to solve real-world research challenges.
Schrödinger has a strategic partnership with NVIDIA to optimize our computational drug discovery platform for NVIDIA GPU technology.
Get answers to common questions and learn best practices for using Schrödinger’s software.
Learn more about the related computational technologies available to progress your research projects.
Fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement
A modern, comprehensive force field for accurate molecular simulations
Accurate ligand binding mode prediction for novel chemical matter, for on-targets and off-targets
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.