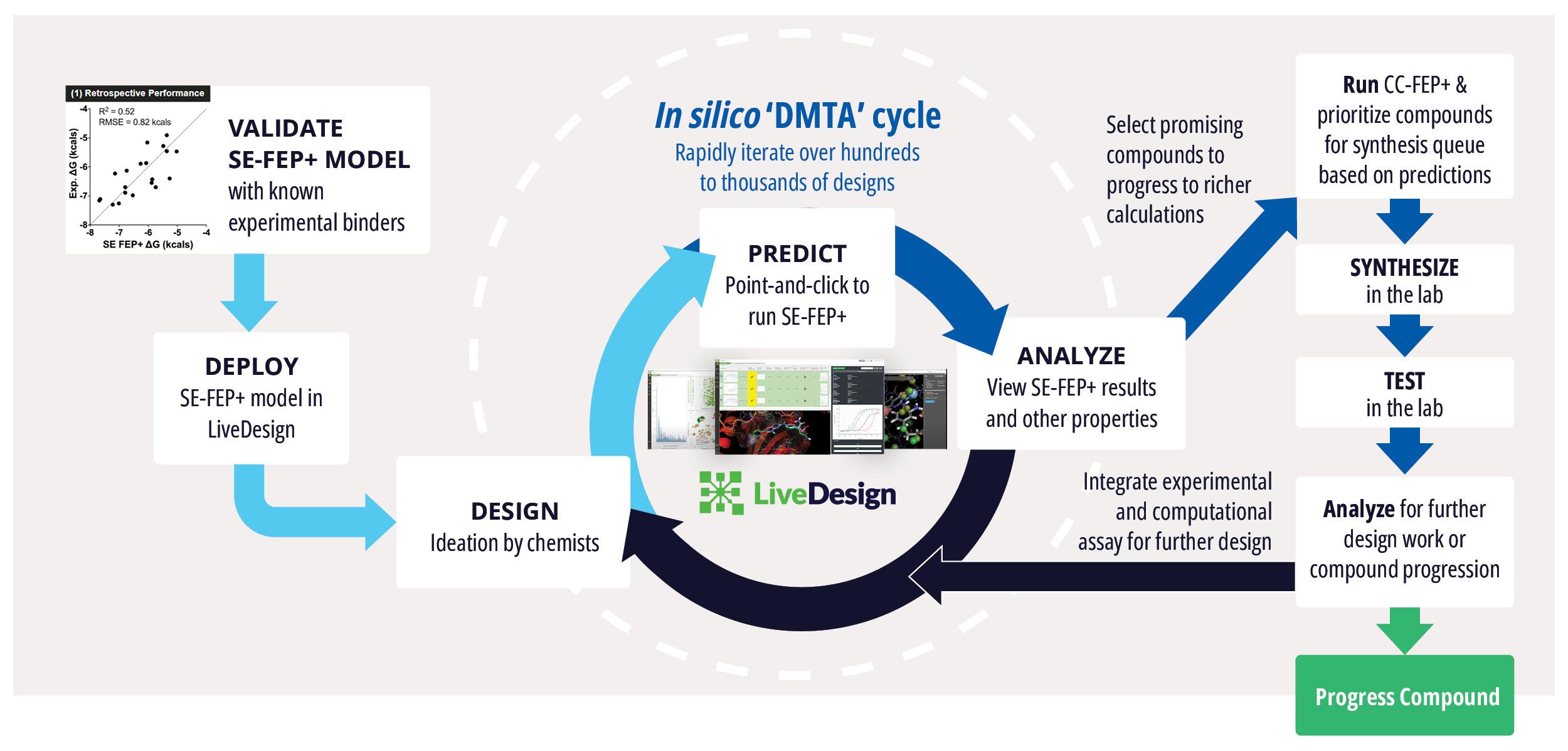
Accelerating DMTA cycles with fast, push-button free energy calculations available to whole project teams
Single-edge FEP+ integrated in LiveDesign
Executive Summary
FEP+ is a widely adopted technology for accelerating small molecule drug discovery programs, with application in hit discovery, hit-to-lead, and lead optimization. The accuracy and utility of FEP+ as a computational assay for the prediction of binding energies is validated extensively, with predictions falling within 1.0 kcal/mol of experimental values on average.1,2 With accuracy approaching lab experiment, FEP+ serves as a digital binding affinity assay for both on- and off-targets which enables drug discovery teams to efficiently optimize molecular profiles and pursue novel chemistry with confidence.
Given the computational cost and complexity of running free energy calculations, adoption is largely driven by expert computational chemists who then manually share results with the project team. To amplify the impact of these methods and improve the agency of individual team members, they should be accessible directly to medicinal chemists and be able to be run at a scale comparable with traditional virtual screening methods.
Toward these goals, developments in expanding the accessibility of free energy calculations — in particular single-edge FEP+ (SE-FEP+) — enable a new paradigm in structure-based molecular design. SE-FEP+ simulations can be run in a fraction of the time of full cycle-closure FEP+ (CC-FEP+) simulations and can be conceptualized as ‘push-button’ for execution. This makes it easily deployable in LiveDesign, a cloud-native, collaborative enterprise design platform. This workflow enables all project team members to integrate FEP+ within their design workflows, resulting in a highly interactive and fully in silico design-make-test-analyze (DMTA) cycle where chemists are empowered to digitally test hypotheses and iteratively improve designs prior to compound synthesis (Figure 1).
In one such oncology program within Schrödinger’s Therapeutics Group, the project team was able to improve compound potency over 100-fold by running many iterative in silico DMTA cycles using FEP+ in LiveDesign over the course of a four week period.
Single-Edge FEP+:
Accurate prediction of relative binding affinities in a fraction of the time
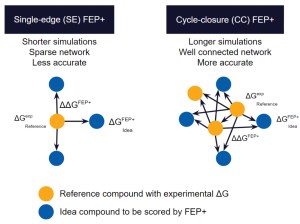
As compared to docking or MM-GBSA calculations, full cycle-closure FEP+ (CC-FEP+) simulations enable highly accurate rank-ordering of compounds, but are compute intensive and complex to set up, reducing their ability to be set up and executed by non-expert molecular modelers.
Single-edge FEP+ (SE-FEP+) simulations have been shown to maintain high predictive accuracy but can often be run ~10x faster than CC-FEP+ simulations in an automated fashion to triage new designs efficiently. Once the SE-FEP+ model is validated, it can be used in a push-button fashion to test new designs that share a common scaffold (i.e., common core or common R-group) with the reference compound and enable efficient scoring of large numbers of idea compounds (Figure 2).
When deployed via a collaborative platform such as LiveDesign, SE-FEP+ gives project teams the ability to run rapid design cycles and prioritize ideas with confidence. The most promising design ideas can then be run through full CC-FEP+ calculations to obtain the most robust potency predictions for informing synthesis queue decisions. If a compound falls outside the criteria established by the computational chemist for running the SE-FEP+ models, that molecule can be added to a queue for running full CC-FEP+ in a more manual setup by a computational chemist.
![]()
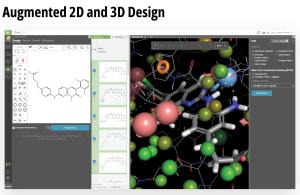
A complete digital molecular design lab
LiveDesign is a flexible, cloud-native collaborative working environment for entire discovery teams. The platform democratizes digital design processes and enables more productive design cycles by harnessing the power of medicinal chemistry strategies, advanced cheminformatics, cutting-edge computational chemistry workflows, virtual ‘design and test’ technologies, and centralized access to live project data — all in a single interface.
The platform empowers creativity by democratizing access to powerful predictive modeling workflows, such as push-button free energy calculations, and allows teams to efficiently capture and progress their most promising design ideas. LiveDesign also centralizes team collaboration and decisionmaking — enabling crowdsourcing of design ideas, interactive team-based design, and easy sharing of data with internal and external partners.
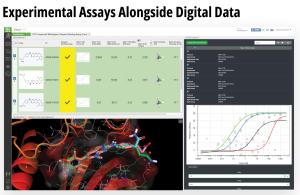
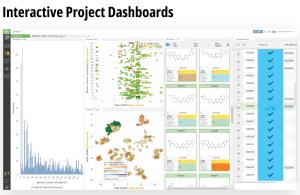
Case Study:
Oncology Program SCH-01
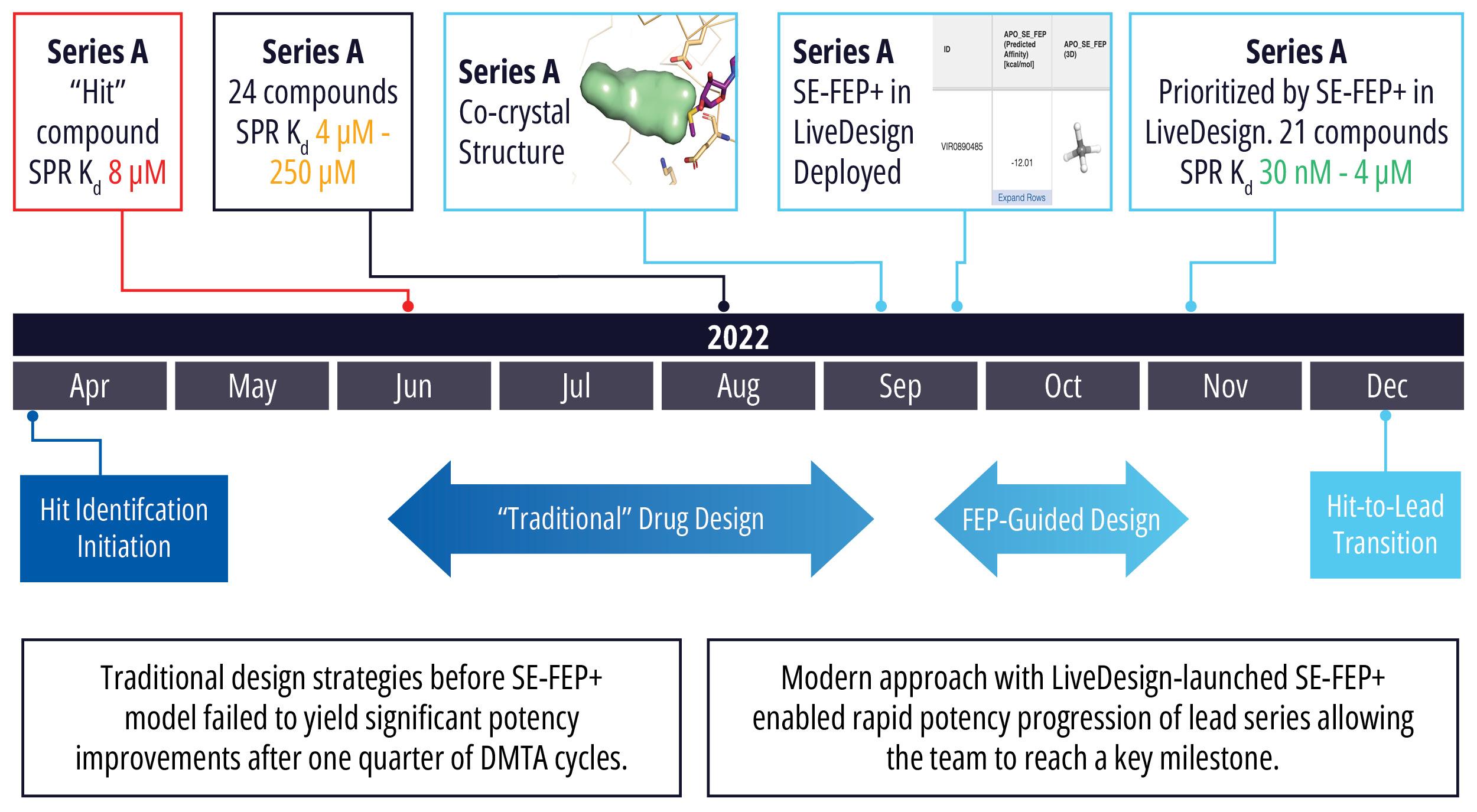
In an early stage oncology drug discovery program driven by Schrödinger’s Therapeutics Group, the project team faced a design challenge to find and optimize an initial hit series. Of the 200 compounds synthesized in the program to date, the most potent molecule had a modest binding affinity of 2.7 μM, which failed to meet the criteria for the hit-to-lead transition. Traditional design strategies had failed to yield significant potency improvements after several months of DMTA cycles, even in the most promising series.
With a new cocrystal structure for this promising series in hand, the team turned to FEP+ to address the potency challenge. An SE-FEP+ model was developed and validated retrospectively using known experimental binding affinities. The SE-FEP+ model was then deployed in LiveDesign, allowing medicinal chemists to design new ideas and independently run SE-FEP+ calculations with the click of a button.
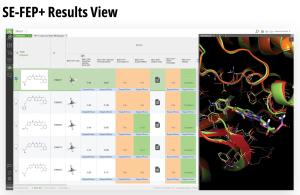
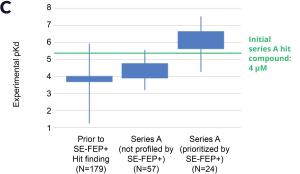
Using this validated model, the project team iteratively designed molecules and digitally assessed their ability to improve potency from the current best Series A potency of 4 μM. During one particular four week period, approximately 400 designs were profiled with medicinal chemistry team members contributing and running SE-FEP+ in LiveDesign for ~65% of the designs. No new compounds were experimentally synthesized and tested in this month, but the team was able to significantly optimize compound potency in silico as predicted by FEP+. Each week, the predicted potency of the designs made by the chemistry team improved until the top scoring compounds were then prioritized for synthesis and experimental profiling (Figure 3).
By prioritizing 24 compounds for synthesis based on SE-FEP+ predictions in LiveDesign over this one month period, the project team dramatically enriched the synthesis queue with potent designs. In fact, 21 compounds exhibited improved measured binding affinities of less than 4 μM (Figure 4). Based on this significant progress, the team moved the program forward from the hit identification stage to the hit-tolead stage in a highly accelerated time frame.
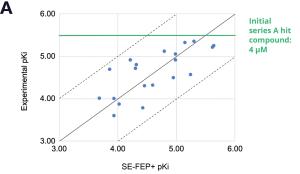
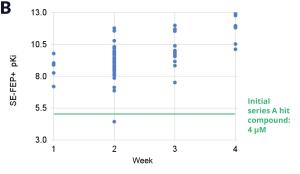
Conclusion:
Deployment of SE-FEP+ in LiveDesign presents a new paradigm for computationally-guided design by democratizing access to powerful, push-button FEP+ calculations to teams of medicinal chemists. It enables chemists to think broadly and creatively when designing ideas by lowering the barrier to in silico testing of ideas before synthesis. Additionally, the workflow enables expert computational chemists to create guardrails around the use of FEP+ by chemistry teams, and focus time and effort on the more complicated full-cycle challenges to the highly triaged compounds.
This workflow can assist in rapid potency progression, optimization of selectivity against known off-targets, and maintenance of potency while optimizing other physicochemical properties. This allows project teams to quickly reach key milestones and design the best possible molecules to meet the target product profile while dramatically reducing the number of compounds synthesized and tested.
![]()
How it works
Succcesful deployment of SE-FEP+ in LiveDesign follows these key steps:
- A scientist trained in FEP+ creates and validates an FEP+ model on a specific protein-ligand series of interest.
- A model execution strategy is determined around permitted structural modifications, licenses, and available compute hardware as these calculations are computationally significant.
- The FEP+ model is uploaded into LiveDesign and shared via a user-input constrained model.
- In LiveDesign, collaborators design structural modifications to the reference molecule and run SE-FEP+ calculations through the push of a button.
- Results are provided directly in LiveDesign, including the corresponding predicted changes in free energy for each design idea, simulation statistics, and the 3D pose of the ligand in the protein.
- Teams can continue to ideate and explore modifications to identify promising compounds. The most promising compounds are then progressed and further scored with CC-FEP+ for the highest accuracy binding affinity prediction.
![]()
Best Practices
Key points to consider before deploying SE-FEP+ in LiveDesign:
- Ensure your SE-FEP+ model is well-validated using known experimental binding affinities.
- Educate all project team members on accurate interpretation of model predictions and to build appropriate confidence in the SE FEP+ model. The Schrödinger Online Course, “Free Energy Calculations for Drug Design”, is a useful resource to learn more about FEP+, including a hands-on case study.
- Develop template reports in LiveDesign that include automated safeguards around what structural modifications can be run successfully using the SE-FEP+ models.
- Ensure sufficient cluster compute resources are secured to run SE–FEP+ simulations efficiently based on anticipated throughput.
- Schrödinger is happy to partner with users to share best practices and provide guidance on optimizing FEP+ workflows for discovery programs. Reach out to your local Schrödinger team for assistance.
References
-
Impacting Drug Discovery Projects with Large-Scale Enumerations, Machine Learning Strategies, and Free-Energy Predictions
Knight et al. ACS Symposium Series, 1397, 2021, 205-226
-
Advancing Drug Discovery Through Enhanced Free Energy Calculations
Abel et al. Acc. Chem. Res. 2017, 50, 7, 1625–1632
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.