20 years of Glide: A legacy of docking innovation and the next frontier with Glide WS
Overview
Glide has long set the gold standard for commercial molecular docking software due to its robust performance in both binding mode prediction and empirical scoring tasks, ease of use, and tight integration with Schrödinger’s Maestro interface and molecular discovery workflows.
To mark the 20th anniversary of the original publication of Glide in 2004, one of the most cited papers in the Journal of Medicinal Chemistry to date1, Schrödinger is proud to introduce Glide WS, a new molecular docking workflow within Glide which leverages water energetics from WaterMap. The release of Glide WS further demonstrates Schrödinger’s commitment to the development of accurate tools that meet the needs of today’s drug discovery teams.
Glide WS offers:
- Enhanced pose predictions by incorporating a flexible description of explicit water molecules
- Efficient scoring and prioritizing of ligands to improve hit enrichment rates
- Reduced false positives by filtering out compounds that are incompatible with the target binding site in virtual screens
- A comprehensive scoring function calibrated by FEP+ calculations and experimental data
Key Features
Glide WS is a molecular docking workflow built on the foundation of Glide SP2, 3 and WScore4 that provides significantly improved sampling and scoring of small molecules in the binding pocket. The Glide WS scoring function was developed with quantitative guidance from experimental data and FEP+ calculations. It uses information from WaterMap5 to evaluate the water energetics associated with desolvation and capture water mediated interactions. Additionally, Glide WS incorporates MM-GBSA calculations to assess the overall goodness of fit of the ligand pose in the binding site.
Enhanced conformation generation for pose prediction
Glide WS computes the ligand conformer distribution using a novel approach that combines an RDKit algorithm and ConfGen. By focusing on ‘hard to sample’ ligand features such as non-aromatic rings, this new method realizes substantial gains in the generation of native-like ligand conformers while keeping computational costs within reach for most practical docking and virtual screening applications. As shown in Figure 1, improved ring sampling in Glide WS predicted the correct conformation of the 7-member ring in the middle of the molecule.

Scoring function calibration guided by FEP+
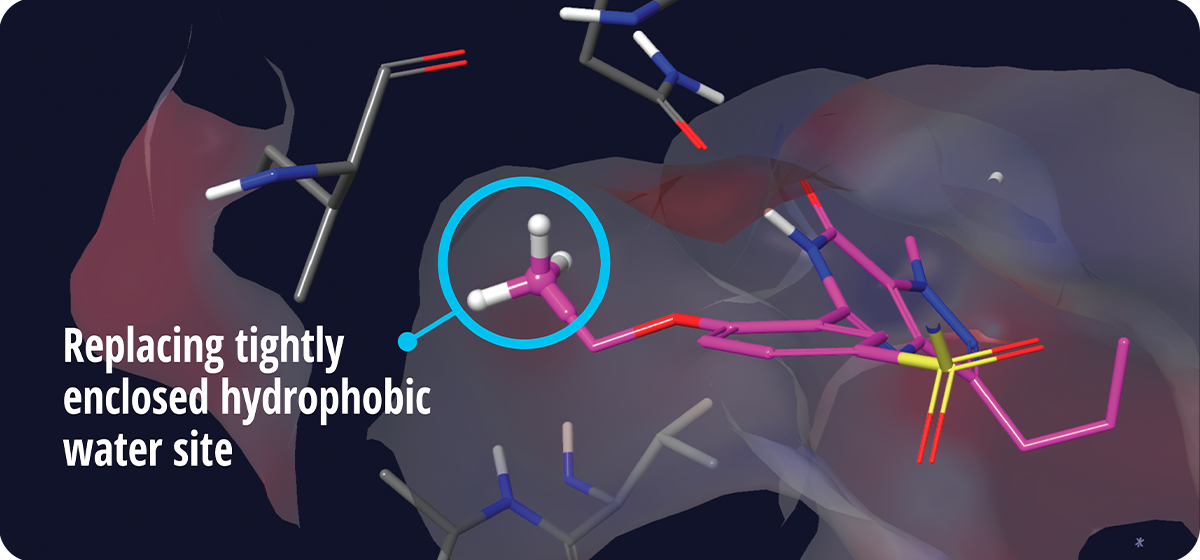
The Glide WS scoring function is calibrated by FEP+6, a widely established benchmark computational method for binding affinity prediction, to yield more comprehensive reward and penalty terms. The scoring function in Glide WS is more robust than Glide SP, with many additional scoring terms based on a detailed study of thousands of PDB structures. As an example, Glide WS can identify “magic methyl” sites in good agreement with FEP+ and experimental data, wherein the addition of a single heavy atom can provide a seemingly nonintuitive boost in the potency of the ligand (Figure 2). By accurately capturing this important effect, Glide WS is able to achieve better separation of true ligands from false positives across many targets.
Superior performance on self-docking
In a curated protein-ligand dataset with 765 PDB complexes, Glide WS is able to reproduce the ligand pose for 98% of the cases compared to 88.7% for Glide SP and 91% for Glide XP7 (Table 1). However, Glide WS is notably slower than Glide SP due to the extensive sampling and more comprehensive scoring of the poses. Glide WS is best used to refine and filter a subset of compounds from Glide SP screening as discussed in the section below.

Outperforms Glide SP on key virtual screening benchmark
A common use case of molecular docking is in silico screening of real and virtual compounds to identify novel hits. As the number of compounds experimentally tested will typically be much less than the number screened, it is vital that a docking method be able to recover true binders near the top of the docking output— this is known as early enrichment. We compared the early enrichment performance of Glide WS and Glide SP on a diverse subset of 23 targets from DUD-E. DUD-E is a popular benchmark designed to evaluate the performance of docking tools (Figure 3). For each target, DUD-E provides a set of diverse ligands and property-matched decoys.
As Glide WS uses the difference between experimental binding affinity and docking score of the native ligand to estimate reorganization energy, the Glide WS docking score is a more realistic estimation of the true binding affinities across different targets. Here, we applied a cutoff based on the Glide WS docking score (-8kcal/mol) and selected up to 100 ligands to mimic a real virtual screening campaign and considered the same number of ligands screened by Glide SP to have a fair comparison.

Both Glide WS and SP achieve reasonable hit rates for the targets investigated, with Glide WS finding more active compounds as shown in Figure 4a. This type of analysis does not consider the potential for the competing decoys to be active and potentially underestimates the value of rescoring top hits.
We further investigate the decoys using absolute binding free energy perturbation (ABFEP)8. ABFEP provides a theoretically more rigorous and accurate description of protein–ligand binding thermodynamics, predicting the binding affinities with errors on the same order of magnitude of experimental variations. This investigation helps us better understand the nature of the high-ranking decoys and identify clear false positives coming out from the Glide WS and SP screens, i.e., decoys with good docking scores but bad ABFEP scores. As shown in Figure 4b, Glide WS generates much fewer bad decoys (ΔGABFEP+ greater than -7kcal/mol) than Glide SP among the top scored ligands. Thus, we find, based on the ABFEP scoring data that the Glide WS decoys are much more likely to be active compounds than the Glide SP decoys. This in turn substantiates that Glide WS is a more effective filtering tool in a virtual screening campaign for selecting compounds to be purchased or to be rescored with ABFEP within the context of a full virtual screening funnel.
When to use Glide WS
Glide WS is a more advanced docking tool than Glide SP, providing better poses and fewer false positives, while still being much less expensive than ABFEP+. As Glide WS is ~20x slower than Glide SP, we recommend using Glide WS to further filter virtual hits from a Glide SP screening. Glide WS can also be used in a lead optimization stage to help filter ideas coming from enumeration or other ideation tools.
License Requirements: Glide, WaterMap
References
-
Research.com. (n.d.)
Journal of Medicinal Chemistry: What are the most cited papers published in the journal? Retrieved on October 28, 2024, from https://research.com/journal/journal-of-medicinal-chemistry.
-
Glide: A new approach for rapid, accurate docking and scoring.
1. Method and assessment of docking accuracy. Friesner et al. J. Med. Chem. 2004, 47, 7, 1739–1749.
-
Glide: A new approach for rapid, accurate docking and scoring.
2. Enrichment factors in database screening. Halgren et al. J. Med. Chem. 2004, 47, 7, 1750–1759.
-
WScore: A flexible and accurate treatment of explicit water molecules in ligand–receptor docking.
Murphy et al. J. Med. Chem. 2016, 59, 9, 4364–4384.
-
Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding.
Young et al. Proceedings of the National Academy of Sciences, 104, 3, 808-813.
-
Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field.
Wang et al. J. Am. Chem. Soc. 2015, 137, 7, 2695–2703.
-
Extra Precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes.
Freisner et al. J. Med. Chem. 2006, 49, 21, 6177–6196.
-
Enhancing hit discovery in virtual screening through absolute protein–ligand binding free-energy calculations.
Chen et al. J. Chem. Inf. Model. 2023, 63, 10, 3171–3185.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Modeling Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.