The new solution to the induced fit docking problem: How IFD-MD rapidly and reliably predicts accurate ligand binding
Scientists from Schrödinger’s Structure Prediction, Structure Refinement, and Drug Discovery Groups recently published Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein-Ligand Binding in The Journal of Chemical Theory and Computation. The authors found that Schrödinger’s IFD-MD is able to obtain an accurate structure to use in structure-based drug design (SBDD) more quickly and easily than experimental determination.
Dr. Ed Miller, Product Manager Prime Structure Prediction Group, describes our next generation induced fit docking workflow which provides an accurate and reliable way to determine binding modes of novel ligand scaffolds.
Explain what this paper means. Why is it so exciting?
It’s fairly common in drug discovery for researchers to have access to a structure of their target of interest, or of a close homolog. And yet despite these pre-existing structures, a single side chain, a few atoms even, can render that structure useless for your particular drug discovery project. Obtaining an accurate characterization of the protein-ligand binding mode for your particular compound series is a key starting point for rigorous structure-based modeling efforts, such as free energy methods, to accurately predict affinity. Structures in complex with other ligand scaffolds, not of interest to the drug discovery program, have limitations for scientists designing novel scaffolds.
Historically, significant time and money has been spent to overcome this challenge by obtaining new experimental x-ray crystal structures bound to a team’s own small molecule. This can cause project timelines to extend and delay. For some proteins such as membrane proteins or GPCR’s, obtaining an experimental structure is extremely difficult, if not impossible. There has been a real need for computational methods that can solve this issue in a fraction of the time.
What this paper shows is that by using a new technology called IFD-MD we can produce very highly quality structures and predict protein-ligand binding modes reliably and efficiently for these exact types of problems.
However, for a method like IFD-MD to impact projects, people have to trust the computational models as much as they trust an experimentally-derived crystal structure. They need to have confidence they can use the model prospectively to guide predictions, even in the absence of an experimental structure. That’s a tall ask, but what’s exciting is by challenging and validating the models upfront with free energy calculations (FEP+), scientists can confidently use structures produced by IFD-MD to impact active projects. For example, if FEP+ results using the IFD-MD produced structural model show strong correlation retrospectively, the scientist can have confidence in using the model prospectively. And indeed in the paper, we show this to be the case, prospectively, for two separate internal programs.
What is the difference between Schrödinger’s IFD software (Induced Fit Docking) and IFD-MD?
More than a decade ago, Schrödinger introduced an automated induced fit docking (IFD) algorithm, based on combining our Glide docking program with our Prime protein modeling program. The Glide/Prime IFD methodology has been used extensively in drug discovery projects in the pharmaceutical and biotechnology industries up to the present day. However, limited sampling sometimes means IFD fails to identify or accurately rank native-like protein-ligand binding poses.
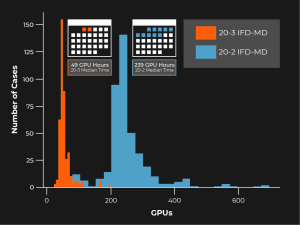
Five years ago, Schrödinger embarked on a project aimed at improving the accuracy and reliability of IFD. The result of these efforts is IFD-MD (Induced-Fit Docking Molecular Dynamics). The method is computationally much more efficient than brute-force molecular dynamics (MD) simulations, and can easily be completed overnight using modest cloud computing resources. In fact, we’ve even made some significant improvements in speed in recent product releases (Figure 1).
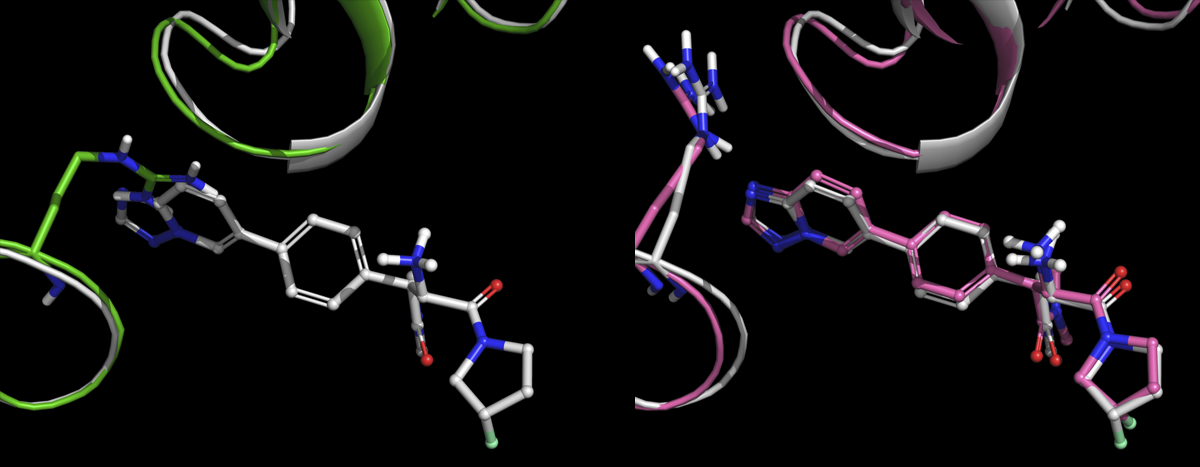
We also found that a very high fraction (90% or better) of both training set and test cases reproduce key features of the crystal structure, and yield molecular dynamics trajectories that are very similar to those obtained when starting from the crystal structure.
We subsequently tested the performance of IFD-MD for five proprietary systems investigated in the course of Schrödinger’s in-house drug discovery efforts and asked the question:
“Could one have predicted a proprietary structure with an accurate protein-ligand binding pose using only the publically available structures that were present at the time the drug discovery project was initiated?”
The results of our cross-docking experiments (i.e. docking with a different small molecule than the original experimentally-bound small molecule) are described in the chart below (Figure 2). We compared results across rigid receptor docking method (GlideSP), our original IFD method, and our new IFD-MD method.
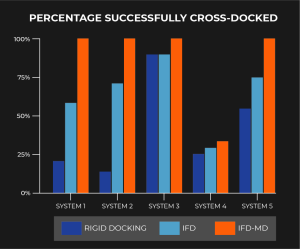
What was unique about this validation is that protein-ligand systems were not eliminated based on presence or absence of backbone motion or if the template ligand fully occupied the binding site of the target molecule. In fact System 4, the only system that did not consistently produce a successful cross-docked complex 100% of the time, does require significant backbone reorganization which is currently beyond the scope of this method. However, even in prospective cases where severe backbone motion may be unknown but present, testing and validating IFD-MD models using FEP+ de-risks this limitation. Most excitingly, IFD-MD outperforms in all five systems compared to GlideSP and IFD.
Can you describe the IFD-MD workflow?
Years of development concluded in the IFD-MD workflow summarized in Figure 3. IFD-MD integrates multiple Schrödinger tools into a single solution for predicting binding poses, and operates as a series of coupled CPU and GPU jobs.
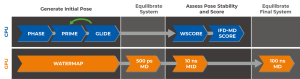
Initial pose generation is done using pharmacophore docking (Phase) followed by structure refinement using Prime and redocked with Glide in an iterative process. Hydration sites are estimated by calculating their thermodynamics properties (WaterMap) and used for water placement in the binding pocket. After a short system equilibration the stability of the ligand poses are assessed using metadynamics (MtD) simulations and scored.
You note that the IFD-MD method is computationally much more efficient than brute-force MD simulations. Can you explain that?
Perhaps the most straightforward approach to induced-fit docking is to simply simulate, with MD, the ligand traveling from solution into the binding site, with all the atoms in the system, the receptor, the ligand, the solvent, free to move. This is the brute-force approach. There are groups which have shown that it can be done, D.E. Shaw Research most famously. But to do this reliably is very time consuming and expensive, and without a large number of simulations across a range of systems, it’s not clear precisely what is necessary and sufficient to get reliable results.
In an active project, the uncertainties one has to consider start to escalate. There’s alternative tautomeric states, stereoisomers, alternative ligand series, and lots of other variables. Brute force MD simulations are just too expensive to be practical here because each of these variables require a separate simulation, not to mention the valuable time spent by the scientist in having to analyze the large volumes of data generated by brute force MD.
In a recent publication authored by scientists at Merck KGaA, Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects, they found that FEP+’s applicability could be limited by a lack of structural data for a target of interest. Do you see IFD-MD having any effect on that?
Absolutely. In that publication, the authors state that the main reason limiting the use of FEP+ in some of their discovery projects is a lack of structural data. In particular, protein conformational changes, unresolved atoms in an existing crystal structure, or uncertainty in the binding exist as possible challenges. The induced fit docking algorithm we present in this publication is intended to significantly improve these issues in active drug discovery projects.
Any final thoughts?
Endemic in many computational methods is the rigid-receptor approximation. This approximation has historically shown great utility, but by stepping past it with induced fit methods, a whole realm of new, more accurate simulations become possible. In the near future, we’ll be pushing the limits further of how IFD-MD can impact active drug discovery projects.
Author

Edward Miller
Senior Director of Protein Structure Modeling
Edward Miller joined Schrödinger in 2014, and is responsible for advancing the domain of applicability of structure-based drug discovery into challenging targets and off-targets. Dr. Miller obtained his PhD from Columbia University, where he was awarded a DOE research fellowship. His thesis work with Professor Richard Friesner involved developing methods to accurately model loop conformations across a broad array of protein families. His recent work has been focused on methods development for induced fit docking and protein structure refinement.