Glide
Industry-leading ligand-receptor docking solution

Industry-leading ligand-receptor docking solution
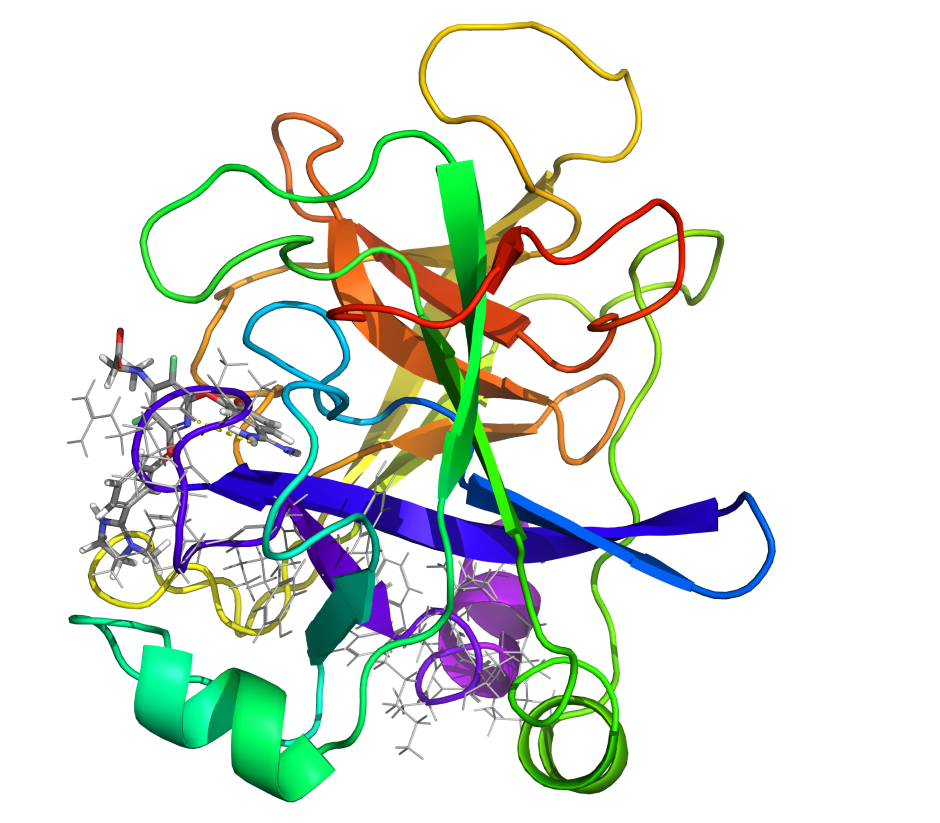
Glide is the leading industrial solution for reliable ligand-receptor docking. It augments and accelerates structure-based drug design across a range of applications, including virtual screening, binding mode prediction and interactive 3D molecular design.
Easily create and validate docking models with a simple, guided graphical user interface
Achieve high enrichment across a diverse range of receptor types, including small molecules, peptides, and macrocycles
Benefit from a broad range of constraints to easily bias docking calculations to meet experimentally-observed requirements and desired chemical space
Achieve accurate pose predictions and eliminate false positive virtual hits with Glide WS

Glide SP is a widely used and precise docking workflow designed for high-throughput virtual screens. Glide SP employs hierarchical filter technology that is ideal for large-scale screening to yield fast and accurate hits.
Glide WS is an advanced docking tool that leverages explicit water dynamics from WaterMap. Built on the foundation of Glide SP and WScore, Glide WS provides significantly improved sampling and scoring of small molecules in the binding pocket.
Interactively design and dock in 3D using goal-directed ligand design workflows in Ligand Designer and LiveDesign
Accurately predict ligand poses to understand interaction with receptor and provide initial pose for rescoring with AB FEP+
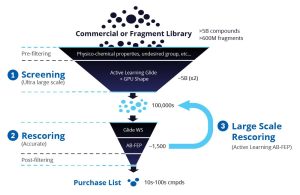
Perform virtual screens with automated workflows that are customizable to fit project needs and accelerate screening of ultra-large libraries (>1B compounds) using Glide enhanced by Active Learning
Dock a set of ligands that bind covalently to the receptor, using predefined or custom reaction chemistry – CovDock
Incorporate detailed water analysis from WaterMap calculations to evaluate protein-ligand binding interactions and reduce false positives from Glide SP screenings
Schrödinger has partnered with leading providers to help you access commercial databases of fragments, lead-like, near drug-like, and drug-like compounds ranging from millions to billions of compounds encompassing a vast chemical space.
Discover how Schrödinger technology is being used to solve real-world research challenges.
Get answers to common questions and learn best practices for using Schrödinger’s software.

Learn more about the related computational technologies available to progress your research projects.
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.