IFD-MD
Accurate ligand binding mode prediction for novel chemical matter, for on-targets and off-targets

Accurate ligand binding mode prediction for novel chemical matter, for on-targets and off-targets
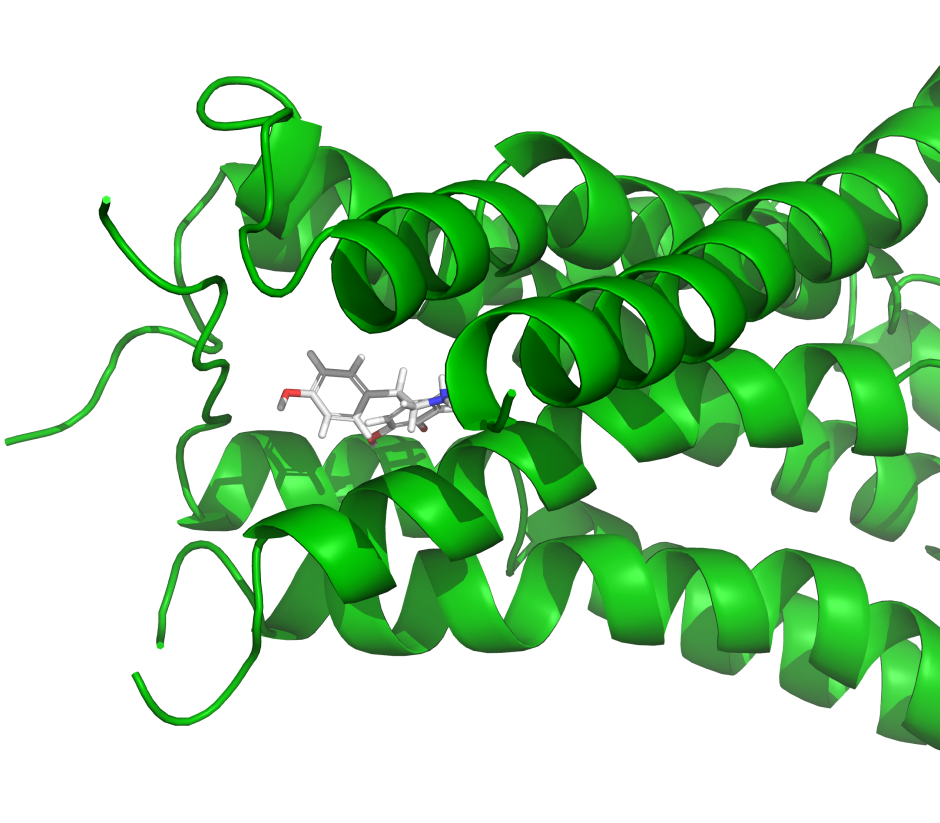
IFD-MD is a powerful GPU-accelerated solution for predicting receptor-ligand binding poses at an accuracy approaching experimental methods, but at a reduced cost and faster turnaround. Using IFD-MD in combination with FEP+ for model validation allows a full in silico method for deploying high precision structure-based drug discovery starting from homology models, AlphaFold structures, or experimental structures bound to unrelated chemical matter.
Progress structure-based design efforts without waiting for an experimental crystal structure of a new chemical series
Eliminate the need to initiate a new crystallization program of a known off-target bound to chemical matter
From non-covalent ligands to covalent ligands and macrocycles
with explicit water molecules in the binding site and explicit lipid molecules in the membrane region

Learn how Schrödinger’s digital chemistry platform facilitates efficient multi-parameter optimization of selectivity, cell potency, and toxicity at scale.
read the case studyGet answers to common questions and learn best practices for using Schrödinger’s software.

Learn more about the related computational technologies available to progress your research projects.
State-of-the-art, structure-based method for assessing the energetics of water solvating ligand binding sites for ligand optimization
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.