- Predict antibody structure using a fully guided homology modeling workflow that incorporates de novo CDR loop conformation prediction
- Perform batch homology modeling to accelerate model construction for a parent sequence and its variants
- Identify and prioritize promising leads by modeling and triaging antibody sequences with prediction tools for structure characterization

Antibody Design
Capabilities for structure-based design of antibodies
In traditional IgG or alternative formats
Construct reliable 3D structural models of antibodies directly from sequence
Streamline rational antibody humanization
- Generate humanized antibodies through CDR grafting in conjunction with targeted residue mutations
- Evaluate the percentage of humanness of resulting constructs
Understand and predict antibody-antigen interactions
- Predict antibody-antigen complex structures through ensemble protein-protein docking
- Enhance resolution of experimental epitope mapping data (e.g. mutagenesis or mass-spectroscopy) from peptide to residue level detail
- Identify favorable antibody-antigen contacts through fast protein-protein docking
- Interrogate and analyze predicted protein-protein interactions with an easily accessible graphical user interface
Derisk development by uncovering potential liabilities earlier
- Identify and prioritize promising leads by modeling and triaging antibody sequences with prediction tools for structure characterization
- Highlight potential surface sites for post-translational modification and chemical reactivity
- Detect potential hotspots for aggregation using computational protein surface analysis
Engineer antibodies in silico
- Accurately predict the impact of residue substitution on binding affinity, selectivity, and thermostability
- Rapidly identify mutants with the highest likelihood of meeting project thermostability, binding affinity, and selectivity requirements using MM/GBSA, a fast molecular mechanics-based method
- Refine candidate selection using a systematically validated, free energy-based workflow which provides an accuracy that reproduces experimentally determined relative free energies
Design high-quality biologics with Schrödinger’s cutting-edge software
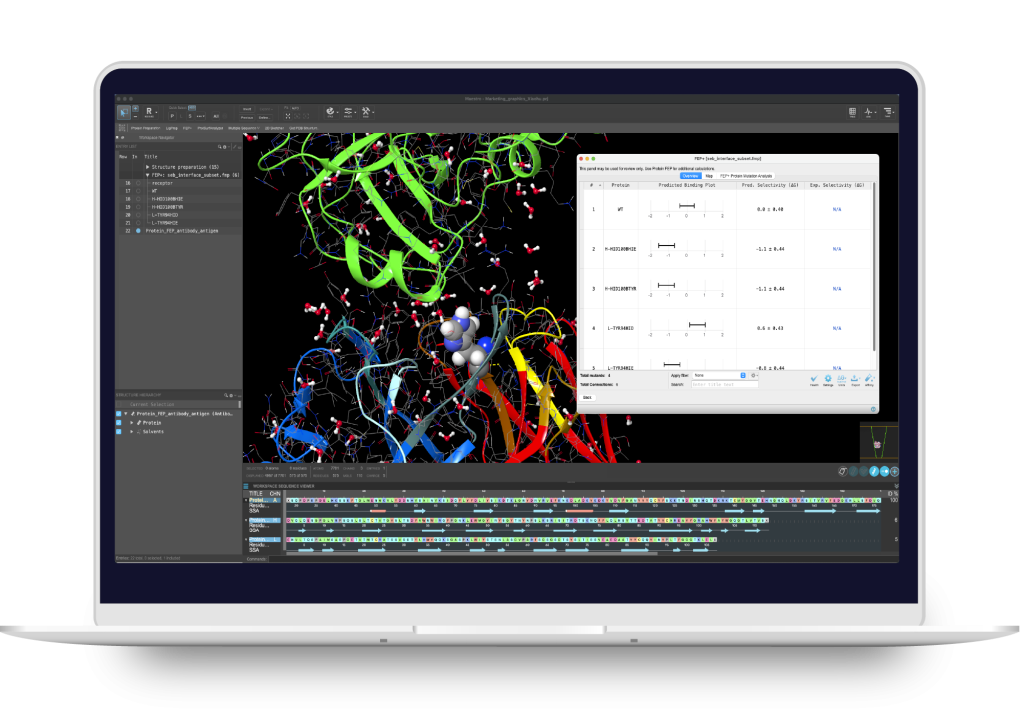
BioLuminate
Modeling environment for biologics discovery
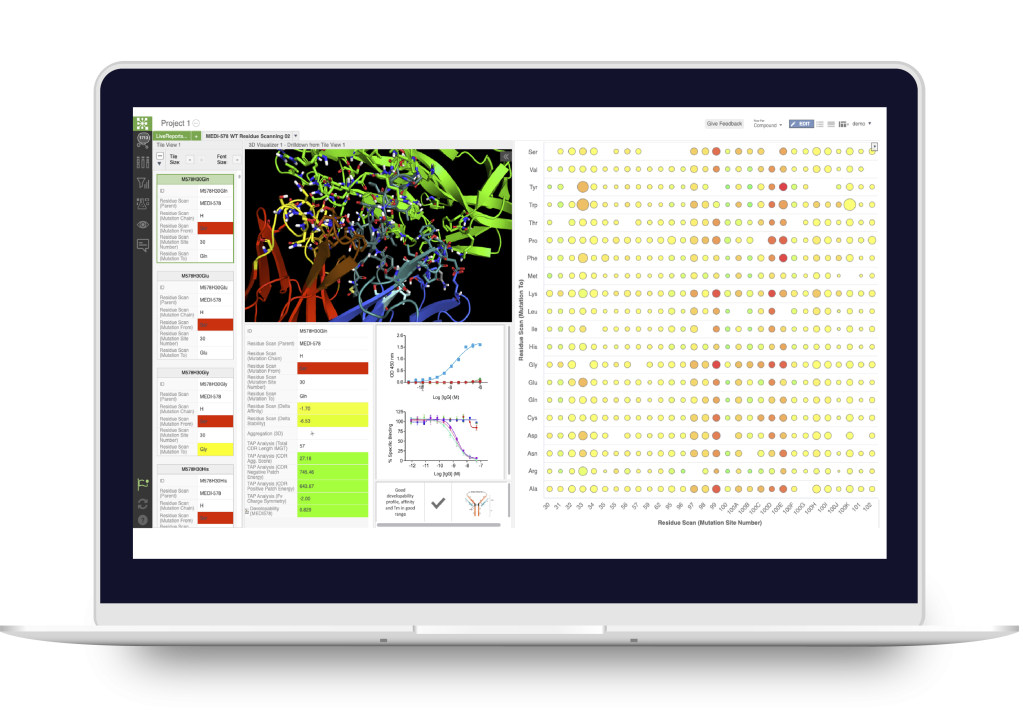
LiveDesign
Collaborative digital biologics design and discovery lab

Featured Course
Learn antibody modeling with our hands-on online course
Level-up your skills by enrolling in our online course, Introduction to Computational Antibody Engineering.
Learn MoreWebinars
Discover how Schrödinger technology is being used to solve real-world research challenges.
Optimizing Protein Stability Using New Computational Design Approaches for Biologics
Accelerating Antibody Drug Discovery Through Computational Modeling
Key Products
Learn more about the key computational technologies available to progress your research projects.
Desmond
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
Publications
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Structure-guided engineering of immunotherapies targeting TRBC1 and TRBC2 in T cell malignancies
Ferrari, M. et al. Nat Commun. 2024
Biological activity validation of a computationally designed Rituximab/CD3 T cell engager targeting CD20+ cancers with multiple mechanisms of action
Cai, W. et al. Antibody Therapeutics, 2021, 4(4), 228-241
Relative Binding Affinity Prediction of Charge-Changing Sequence Mutations with FEP in Protein–Protein Interfaces
Clark, A. J. et al. Journal of Molecular Biology, 2019, 431(7), 1481-1493
Software and services to meet your organizational needs
Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Services
Leverage Schrödinger’s computational expertise and technology at scale to advance your projects through key stages in the drug discovery process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.