Massive theoretical screening of organic semiconductor materials using cloud computing
Panasonic scientists in collaboration with Schrödinger researchers published a paper in The Journal of Physical Chemistry, Massive Theoretical Screen of Hole Conducting Organic Materials in the Heteroacene Family by Using a Cloud-Computing Environment, about performing a large-scale theoretical screen of potential organic electronic materials in a cloud computing environment to determine molecules with optimal hole mobility properties. The performance of organic semiconductor devices depends crucially on the movement of charge through the weakly conducting materials composing the device. Positive charge carriers and negative charge carriers are holes and electrons, respectively. Charge transport can occur by hopping from molecule to molecule through the solid, modeled using atomic scale simulation techniques in the Schrödinger platform. The focus in this work was hole mobility in organic semiconductors.
Panasonic was interested in exploring structures with fused furans, thiophenes and selenophenes. These compounds are interesting for their use as organic semiconductors for a variety of applications. In particular, Panasonic is interested in making printable RF-ID (radio frequency identification) tags that require organic semiconducting materials with charge mobilities higher than currently available compounds. These materials could be used in housing, retail and warehouse management. Noncontact checkout and inventory would further enable future retail transaction technology. For example, a current Panasonic product, called ‘Reji-Robo’ , is a new automated checkout system that cuts labor cost by 10% while speeding up the process for customers.
In this work, Schrödinger created 7,032,432 compounds through structural enumeration as the basis of chemical space for high-performance hole conducting materials. 250,000 of the initial set were randomly selected to perform density functional theory (DFT) calculations of hole reorganization energies. Because of the great amount of compounds to analyze, the team decided to perform the calculations in a cloud computing environment to drastically increase the throughput of calculations.
Leveraging Google cloud computing technology, 3.6M DFT calculations were run to predict hole reorganization energies of the 250,000 compounds—this was completed in just 16 days.

Alexander Goldberg, Senior Principal Scientist within Schrödinger’s Materials Science Group, explains the paper’s key findings:
Can you explain why this work is important?
Obtaining organic materials with improved mobility will help advance the field of organic electronics. Because organic electronic materials are a low-cost, highly flexible, easily processed, lightweight, and sustainable solution there has been a surge in both interest and investment in this field. The global organic semiconductor market alone is expected to grow to $179.4 Billion by 2024, up 22.4% from 2019.1
Although performance of organic material-based devices has improved to the point that it is now challenging inorganic materials for key applications, fundamental research is still needed for further development since the underlying physics is still unclear and working principles need to be clarified. We can utilize the Schrödinger platform to advance this research and enable design of high-quality, next-generation organic electronic materials.
Explain what this paper means. Why is it so exciting?
Performing a screening of this size in this amount of time wasn’t possible even just a few years ago. With the improvements seen in cloud computing, we’ve been able to increase the scale of using atomic scale simulation to screen chemical space drastically. When you consider that screening this number of compounds would take 100 days using a typical 200 CPU HPC resource, you can see just how much faster it is using cloud computing. If you were to screen this number using traditional experimental methods, it would take somewhere around 5,000 years.
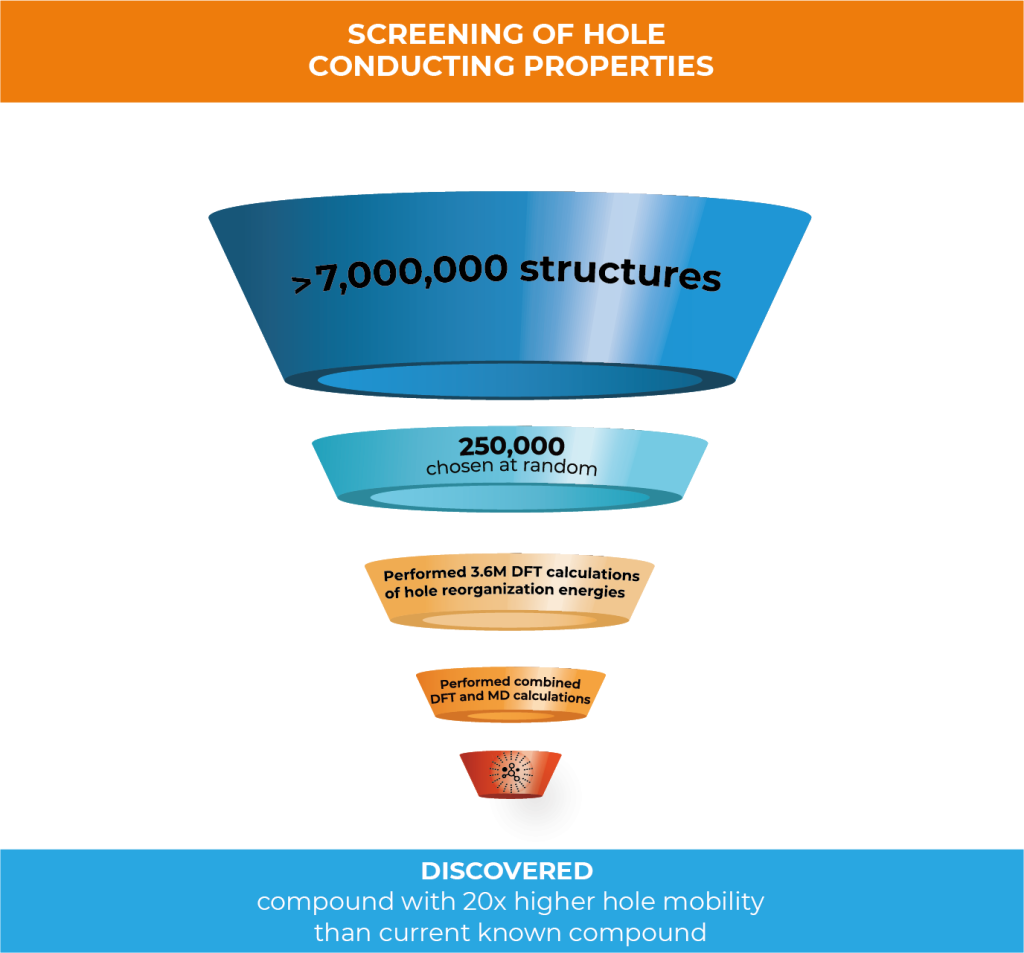
In this project, you evaluated the suitability of 250,000 structures as hole transport materials from a chemical library of 7,032,432 structures to find structures with the most optimal hole mobility. Can you describe the process you used?
250,000 structures were randomly selected to perform density functional theory (DFT) calculations of hole reorganization energies. Then, the hole mobilities of compounds with the lowest 130 reorganization energy were further processed by applying combined DFT and molecular dynamics (MD) methods. By using this method, we were able to identify structures with a predicted hole mobility that is 20 times higher than dinaphthothienophene (DNTT)—a state of the art compound with one of the highest experimental mobilities observed.2
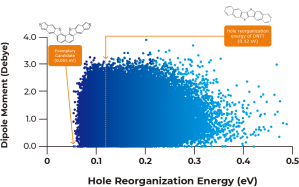
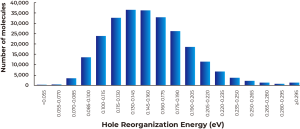
As described in Figure 1A above, calculated dipole moments are plotted against hole reorganization energies for the 250,000 compounds. Good charge transport materials should have small dipole moments to avoid charge trapping in the solid state. The hole reorganization energies range from 0.055 to 0.50 eV. From there, we narrowed down to 17,000 compounds that had hole reorganization energy values below 0.1 eV, as shown in Figure 1b. When you compare that to DNTT which has a hole reorganization energy of 0.12 eV, these 17,000 compounds could all be judged as promising candidates for improved hole transport materials.
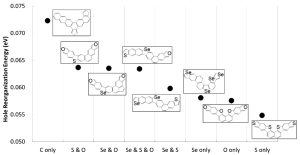
Figure 2 shows the minimum calculated hole reorganization energy in every considered topological compound together with their corresponding chemical structure.
Was there a stepwise selection carried out to filter for high mobility candidates?
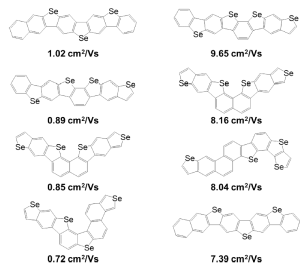
Yes. For example, because the cost of calculating mobility is much higher than calculating reorganization energy, after the massively parallel DFT calculations on the cloud, we selected 130 compounds with the lowest hole reorganization energy for further calculations to obtain estimated condensed phase hole mobility. By calculating 130 compounds we were able to rank which ones had the highest hole mobilities. Figure 3 (below) shows the chemical structures of compounds with the top four highest hole mobilities from that group. Two methods based on different approximations were used to compute mobility in ranking the candidates. Percolation theory is based on a random walk approach, whereas energy disorder theory is based on the distribution of energy states in the solid.
Are there other examples of how Schrödinger uses cloud computing?
Schrödinger utilizes cloud computing not only in Materials Design, but also in our Drug Discovery and Biologics research. Cloud computing enables us to screen vast areas of chemical space in a fraction of the time it would take with on-premise computing resources.
Schrödinger has a history of breaking records for virtual screening in the cloud. In 2013, our materials science team partnered with Cycle Computing, creating a 156,000+ core cloud computing run that was dubbed the Megarun.3 It used virtual machines across eight regions of Amazon Web Servcie’s public cloud around the world for a total of 18 hours and a total cost of just $33,000. It was the largest and fastest cloud computing run at that time in history. When you look at that milestone compared to this screening, you can see how quickly cloud computing technology is advancing.
Earlier this year, Schrödinger partnered with Google Cloud to accelerate our research. With nearly limitless processing power on demand, we are able to explore a larger area of chemical space and model vastly more compounds in a fraction of the time it would take using on-premise computing.
Google also donated resources to advance our work in the fight against COVID-19. Schrödinger is part of a philanthropic initiative with leading biopharma companies from around the world to develop antiviral therapeutics. Google Cloud is providing 16 million hours of GPU time as part of this collaboration. It’s equivalent to accessing the world’s fastest supercomputers.
What’s next?
Our next step is to continue our close collaboration with Panasonic and use the results from this study to enable machine learning techniques to determine structure-property relationships and further screen organic semiconductor design space for high mobility application.
Any final thoughts?
Cloud computing has revolutionized how we do research. Atomic scale simulation is already making a tremendous impact on materials R&D, lowering cost, shortening timelines, reducing risk and driving innovation. Cloud provides essentially, “limitless” resources. We no longer have to limit our pool of candidates to specific libraries. Being able to access the expanse of chemical space means we have a better chance of finding the best candidates with the most optimal properties. And, being able to do it faster means we are able to get results quicker that inform what next steps are needed. It’s an exciting time to be working in materials research.
About the Author

Alexander Goldberg
Ph.D., earned his Ph.D. in Chemical Physics from Tel Aviv University in Israel
Dr. Goldberg has been with Schrödinger for 8 years. His personal research focused on absorption, emission and IR spectroscopy of small clusters. He was also involved in the development of photochemical reactions of organic molecules to be employed in optoelectronic devices as well as finding materials for hydrogen storage in fuel cells and Li battery technology. His research was based on molecular simulations at different length- and time-scales using computational methods of quantum mechanics, classical physics and mesoscale modeling.
References
-
Organic Semiconductor Market Report
Market Research Future; Published September 2020
-
$68 million, 200-year, 150,000-core analytics job run on Amazon’s cloud in 18 hours for $33,000
Brandon Butler; Network World; November 12, 2013