CovDock
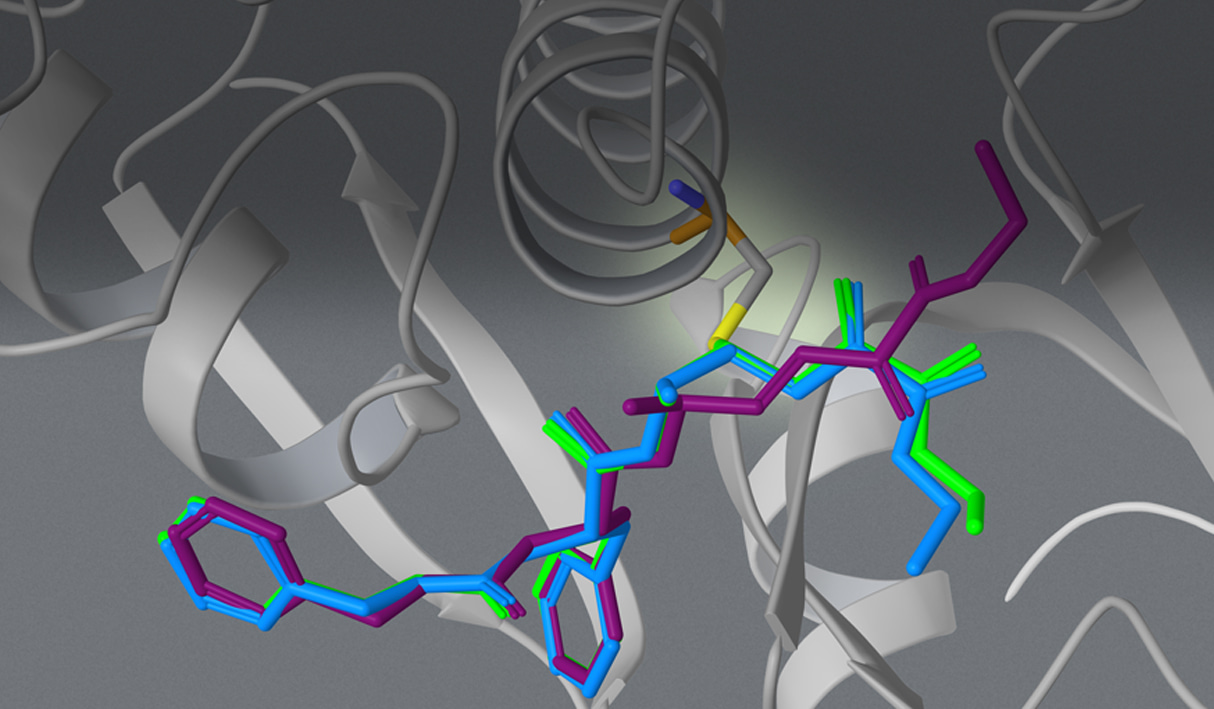
The Advantages of Covalent Docking
With the recent resurgence in covalent drug research, computational insight into covalent docking is becoming key to understanding how covalent inhibitors can be used to address selectivity and potency challenges.
Covalent inhibitors derive their activity not only from the formation of a covalent bond between the target and the ligand but also from stabilizing non-covalent forces in the binding pocket. CovDock selects the top covalent complexes using the extensively validated Prime energy model, and calculates an apparent affinity score that captures these essential elements of a successful covalent docking process:
- The pre-reactive ligand form occupies the binding pocket with enough residency time to facilitate the reaction of the ligand warhead with the reactive protein residue; and
- unfavorable steric clashes and poor electrostatic contacts are prevented as the reaction proceeds.
CovDock begins with Glide docking to a receptor with the reactive residue trimmed to alanine. The receptor reactive residue is then added and sampled to form a covalent bond with the ligand in different poses. Covalent complexes are minimized using the Prime VSGB2.0 energy model to score the top covalent complexes. An apparent affinity score, based on the Glide score of pre-reactive and post-reactive poses, is also calculated to estimate binding energies for use in virtual screening.
Features
Accurate binding mode prediction:
CovDock is built upon a foundation of the time-tested Glide docking algorithm and Prime structure refinement methodology for accurate prediction of non-covalently docked poses. Glide quickly samples a large pool of initial poses for the pre-reactive species and Prime simultaneously optimizes the ligand pose and attachment residue to produce a sound physical chemistry. The resultant accuracy outperforms other docking programs in achieving lower RMS deviations from native co-crystallized structures.
Complete workflow:
CovDock performs a series of automated steps based on a simple setup from the Maestro graphical interface or from the command line. First, CovDock docks the pre-reactive ligand to determine viable poses that bring the reactive group into close proximity with the reactive receptor residue. Then the covalent bond is formed for the top scoring complex structures, the covalently attached ligand is sampled, and the complexes are scored using all-atom molecular mechanics with the OPLS force field and VSGB2.0 implicit solvent model.
Intuitive graphical interface:
Schrödinger’s intuitive graphical user interface, Maestro, provides easy-to-use panels for straightforward set-up of experiments, easy visualization, and efficient analysis of CovDock results.
Covalent Reactions Repository
Schrödinger has made available several custom reactions that can be used in CovDock studies, which can be found on the Covalent Reactions Repository documentation page.
Publications
-
“Docking covalent inhibitors: A parameter free approach to pose prediction and scoring”
Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E., J. Chem. Inf. Model., 2014, 54, 1932−1940.
-
“A Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands”
Toledo Warshaviak, D.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O., J. Chem. Inf. Model, 2014, 54(7), 1941–1950.
Citations
-
Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E., “Docking covalent inhibitors: A parameter free approach to pose prediction and scoring,”
J. Chem. Inf. Model., 2014, 54, 1932−1940.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.