Introducing a new in silico workflow for efficient and automated macrocycle design

Together with Sanofi, Schrödinger recently published a manuscript in the Journal of Medicinal Chemistry, titled “Automated Design of Macrocycles for Therapeutic Applications: From Small Molecules to Peptides and Proteins.” The paper presents the implementation and application of a new in silico workflow for the efficient and automated design of novel and diverse macrocycles.
We sat down with Drs. Andreas Evers (who has since moved to Merck KGaA / EMD Serono) and Michael Wagner of Sanofi, as well as Dr. Dan Sindhikara of Schrödinger to discuss the paper.
Tell us what’s new and key in this paper?
Andreas: In this paper we describe a brand-new workflow to automatically generate, evaluate, and propose synthetic strategies to create macrocycles with desired properties. As you know, macrocycles and cyclic peptides are increasingly attractive therapeutic modalities, and a number of macrocycles with a molecular weight ranging from 500 to 1500 Daltons have become successful drugs, with several examples that can be administered orally. We see macrocyclic molecules as filling an important gap in the world of drugs between small molecules and larger biologics.
As described in our paper in detail, our procedure proved to be very effective, and can be applied for the cyclization of small molecules and peptides and even PROTACs and proteins. We are very excited by the promise shown in our applications of the technology so far.
That’s very interesting, can you tell us how you came to work with Schrödinger on this?
Michael: Within our group at Sanofi, Synthetic Medicinal Modalities, we were interested in coming up with a way to rationally design macrocycles, and we wondered if we could create a method to suggest synthetic strategies for cyclization that would take advantage of well‑established chemical reactions and reagents.
Andreas: At Sanofi, we’ve used Schrödinger’s computational platform for many years on various projects, and while there were tools for studying macrocycles, there was nothing for efficient screening of different attachment sites and possible linkers for a linear starting molecule to automatically design new macrocycles. That’s when we reached out to Dan, and discussed with him what we had in mind, and Dan helped to create this automated workflow, and throughout the project, we worked closely together to validate the new method, as well as to suggest improvements to the technology.
Can you tell us a little more about the workflow?
Dan: Sure, you can find the full details of our integrated approach in our paper, but generally, it involves several steps as this figure illustrates (see below).
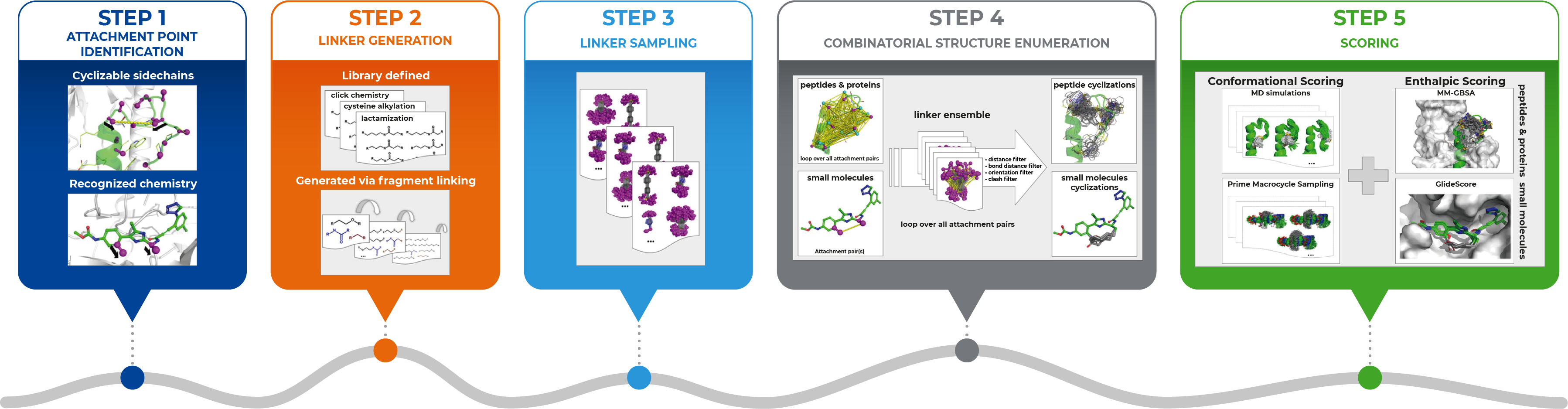
We begin in Step 1, where we use the bioactive conformation of a linear molecule as the starting point – the attachment sites may either be identified automatically or specified by the user. Then in Step 2, we generate the chemical linkers – these may either be provided as explicit lists, e.g., based on available chemical reagents, or optionally, the workflow could generate them automatically by combining smaller fragments. In Step 3, we generate the full conformational ensemble of every linker, and in Step 4, we map the ensemble of linkers to the attachment points, and eliminate unfavorable cyclizations that are not geometrically feasible. Finally, the cyclized ligands can then be ranked through conformational and/or enthalpic scoring. The approach involved using features in various Schrödinger Software including Prime, Desmond, and Glide.
I should also point out that it is possible to generate bicyclic structures in a stepwise manner with our approach, as seen in the retrospective study of the KIX Domain from the Human CREB Binding Protein. We investigated and found that our in silico cyclization approach was able to automatically generate the known bicyclic construct. The case also demonstrates our method’s ability for the engineering of proteins for increased tolerance toward thermal and chemical
denaturation.
Andreas: We put particular emphasis on assuring the synthetic feasibility of the resulting proposals. For this purpose, we compiled an explicit list of linkers based on commercially available chemical reagents. But the list can also be easily modified by the user to consider specific cyclization chemistries and reagents. In addition, we also built in another option that instead of specifying predefined cyclization linkers, the linkers can be constructed in silico from a small set of reactive “breeding fragments,” that can be combined and enumerated to fit within the geometrical constraints of the ligand-receptor complex. As a result, the algorithm will suggest a diverse set of linker chains that are synthetically tractable.
How well did the method perform? What do you see are the key advantages?
Andreas: We carried out several prospective and retrospective studies to validate our approach against different targets and modalities of pharmaceutical interest, and found excellent agreements with experiment. And the workflow automated much of the tedious manual work we used to have to do, and allowed us to more broadly and systematically explore the chemical space for possible attachment sites and linkers.
Dan: Our approach has “synthetic intelligence” built in so that it not only produces novel molecules, but also the list of chemical reagents for their synthesis. In collaboration with the synthetic chemist, the user has the option to reduce or extend the list of reagents, for example, by focusing on a specific cyclization chemistry or considering only reagents that are physically available or can be quickly procured. By doing so, we ensure the resulting design proposals can be straightforwardly synthesized.
Do you think you will use these tools routinely in your drug discovery efforts?
Michael: Definitely, in fact, as we showed in the paper, we used the technology prospectively in two projects – a peptide (dual GLP-1/glucagon receptor agonists) and a small molecule example (TAFIa inhibitors). And in both cases, we were able to design molecules that were novel and highly potent. And the fact that the design proposals can be straightforwardly synthesized is highly appreciated by the medicinal chemists. Therefore, the approach as such is meanwhile used in a standard way in all drug discovery programs where molecules could benefit from cyclization, either in terms of potency, intrinsic stability, or other properties.
Given that until now we’ve never had such a workflow that can propose easily-synthesizable cyclic molecules in an efficient manner before, I believe we can significantly accelerate the cycle times within a drug discovery program.
Besides the demonstrated success in proposing easily-synthesizable macrocycles with desired properties, is there anything else that you find exciting about the potential of this new technology?
Michael: Based on the success in our retrospective studies on systems larger than small molecules and peptides, we believe this approach holds great promise in being applied to other modalities such as PROTACs or even proteins, for example, to improve their thermostability, which is often an important goal in engineering studies for proteins/enzymes for biotherapeutic or biotechnological applications. Currently, we are applying this approach on several internal projects with great initial success.
Authors

Andreas Evers
Principal Scientist Computational and Structural Bioinformatics, Merck KGaA

Michael Wagner
Director Medicinal Chemistry, IDD Synthetic Medicinal Modalities, Sanofi-Aventis

Dan Sindhikara
Principal Scientist, Technical Lead Life Science Software, Schrödinger