Shape-based screening
Shape-based methods for aligning and scoring ligands have proven to be valuable in the field of computer-aided drug design.1,2 Our Shape Screening tool is a powerful shape-based flexible ligand superposition and virtual screening method, which rapidly produces accurate 3D ligand alignments and efficiently enriches actives in virtual screening. Below, we describe the methodology, which is based on the principle of atom distribution triplets to rapidly define trial alignments, followed by refinement of top alignments to maximize the volume overlap. The method can be run in a shape-only mode or it can include atom types or pharmacophore feature encoding, the latter consistently producing the best results for database screening. We show that Shape Screening performs well in database screening calculations when compared with other shape-based methods, including ROCS, using a common set of actives and decoys from the literature. Three key considerations were emphasized when developing Shape Screening:
- The speed at which structures can be processed
- The quality of the superpositions
- The ability to selectively identify actives over decoys within database
Shape Similarity Models
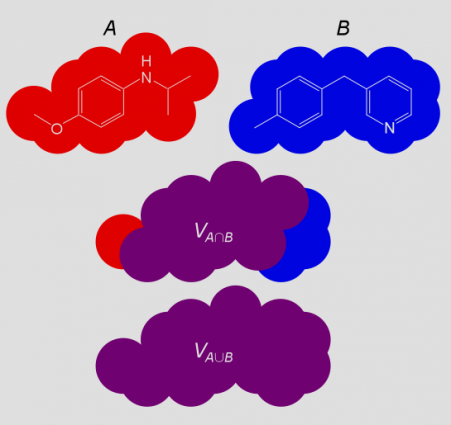
The basic concept of shape similarity is illustrated in Figure 1. Given a superposition of two structures A and B, the shared or jointly occupied volume VA∩B is normalized by the total volume VA∪B to arrive at a shape similarity SimAB that ranges between 0 and 1:
SimAB = VA∩B / VA∪B
A basic goal of shape screening is to determine the alignment of A and B that maximizes SimAB. The complexity of this task depends upon the mathematical representation of shape, and the way in which volumes are calculated.
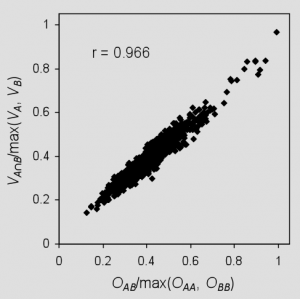
Shape Screening represents a structure as a set of hard atomic van der Waals spheres, with one sphere for each heavy atom and polar hydrogen. The overlap OAB between structures A and B is computed as the sum of pairwise atomic overlaps, and it is normalized by the largest self-overlap to obtain the following measure of shape similarity:
SimAB = OAB/max(OAA, OBB)
This differs from the previous definition in that an alternate normalization scheme is employed, and rigorously computed volumes are replaced by overlaps that ignore the effects of intersections among three or more atoms. Although ignoring higher order overlaps results in an overestimation of the true volumes, normalization by the largest self-overlap computed in the same manner tends to cancel errors, as shown in Figure 2. These approximations allow exceedingly fast shape similarity calculations compared to Gaussian-based methods,3,4 and the use of hard spheres eliminates the need to consider overlap between pairs of atoms separated by more than the sum of their van der Waals radii.
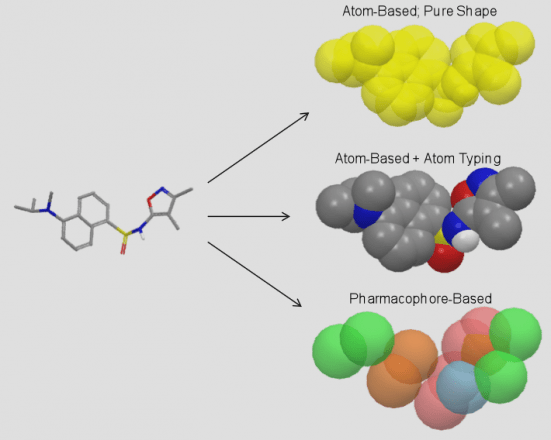
When computing overlaps, Shape Screening has the ability to treat all atoms equivalently, a so-called “pure shape” approach, or to distinguish atoms by type and consider overlap only between atoms of the same type. In the latter case, Shape Screening provides progressively more specific schemes that differentiate by Phase QSAR atom type,5 by element, and by MacroModel atom type.
As an alternative to the atom-based approach, Shape Screening can represent a structure as a set of pharmacophore sites that encode the locations of hydrogen bond acceptors and donors, hydrophobic regions, positive and negative ionizable functions, and aromatic rings. No particular pharmacophore model is implied by this approach, since all sites in a given structure are encoded into the shape, not just those that are hypothesized to be required for binding to a particular target. Pharmacophore sites are mapped to a structure using Phase feature definitions,5 and each site is represented by a 2 Ǻ hard sphere. Figure 3 illustrates the various models of shape that are supported.
Whether an atom-based or pharmacophore-based approach is used, Shape Screening identifies numerous pairs of triplets with similar geometries and similar local environments in structures A and B and superimposes the two structures based on a least-squares alignment of each pair of triplets (Figure 4). The superposition with the highest shape similarity is then refined by realigning on additional pairs of atoms/sites that lie within 0.5 Ǻ of each other in the triplet-based alignment.
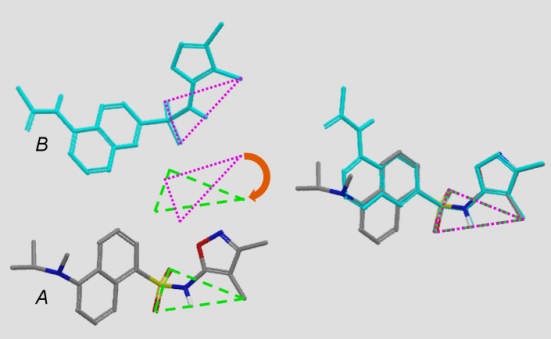
For each pair of structures, hundreds of alignments may ultimately be considered in a tiny fraction of a second. This is possible thanks to an optimized triplet alignment algorithm, ultra-fast hard sphere overlap calculations, and a shape similarity estimation technique which allows poorer overlays to be rejected after computing only a fraction of the total overlap. These time-saving measures allow Shape Screening to screen a multi-conformer Phase database at a rate of about 600 conformers per second on a 2 GHz processor. Shape Screening calculations are trivially parallelizable, and any desired speedup is achievable by dividing the screen over multiple processors.
Shape Screening Applications
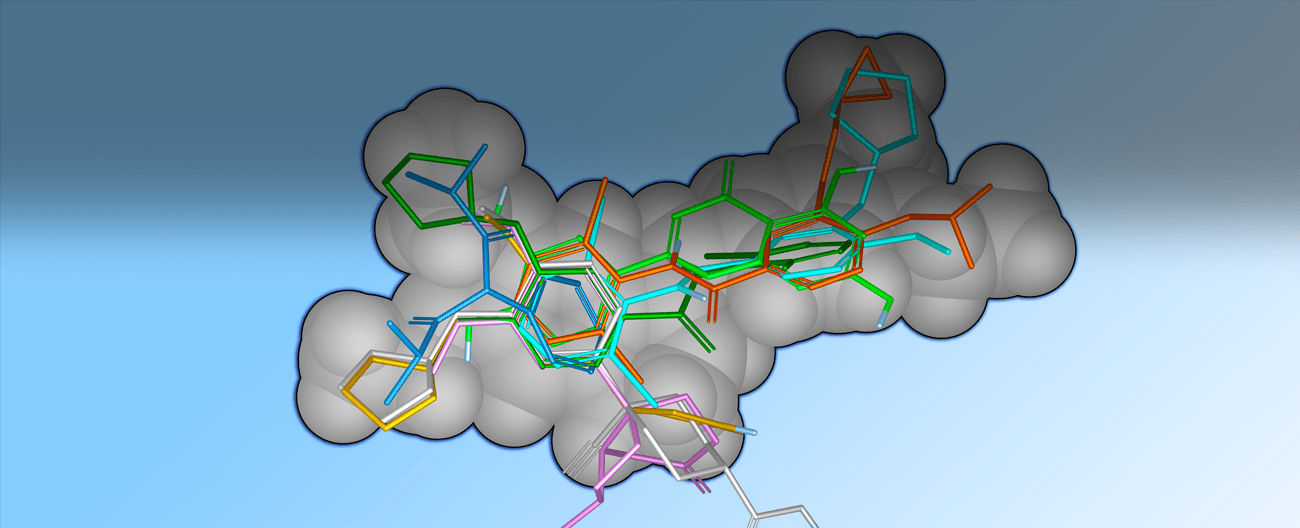
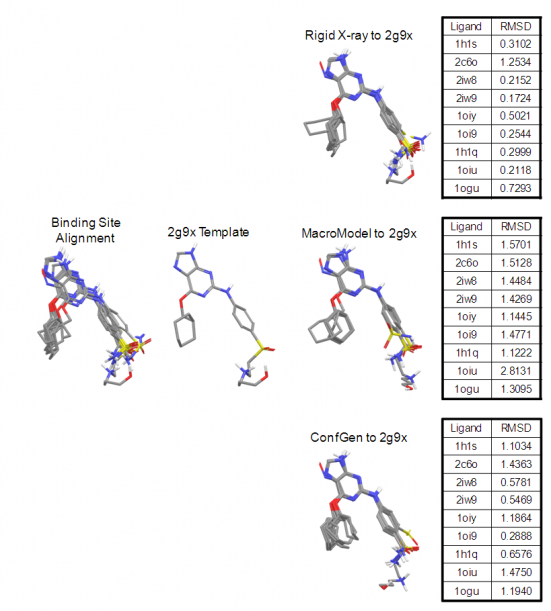
Figure 5 illustrates the quality of overlays that can be achieved with Shape Screening using elemental atom types. Here, the CDK2 X-ray ligand structure 2G9X was used as a rigid template onto which nine other CDK2 ligands were aligned. Results are reported for the highest scoring X‑ray to template alignment, and for the highest scoring conformer to template alignment, where conformational ensembles were generated using both MacroModel and Shape Screening on‑the‑fly ConfGen sampling. In all cases, Shape Screening yields a multi-ligand alignment with low average RMSD values, and clean superposition of common structural elements.
In addition to producing intuitive, high-quality overlays, Shape Screening has been shown to be quite effective at selectively identifying known actives within a database of drug‑like decoys.6 Table 1 summarizes results of Shape Screening virtual screening exercises performed according to the protocols described by McGaughey et al.7 Briefly, multi‑conformer actives for 11 diverse targets were seeded within a multi‑conformer database of 25,000 MDDR decoys. A single active for each target was used as a rigid template for shape-based screening, and database structures were ranked in order of decreasing similarity to that template.
As evidenced by the average enrichment factors in the top 1% of the screened database, results consistently improve with the use of more specific atom typing schemes. Analogous behavior was observed when 2D fingerprint screens were performed on the same data,8 so the relationship between atom type specificity and enrichment is not surprising. Although this trend is promising, improvements for most targets are only incremental, and it is unlikely that devising ever-more discriminating atom typing schemes will lead to a true breakthrough in performance. This threshold for performance improvement is not crossed until the atom-based model of shape is replaced with a pharmacophoric representation. Doing so boosts enrichments for eight of 11 targets, including a two‑fold or greater increase in four cases, and a 66% improvement over MacroModel atom types on average.
| Target | EF(1%) | ||||
|---|---|---|---|---|---|
| Pure Shape | QSAR | Element | MMod | Schrödinger Shape Screening | |
| CA | 10.0 | 25.0 | 27.5 | 32.5 | 32.5 |
| CDK2 | 16.9 | 20.8 | 20.8 | 23.4 | 19.5 |
| COX2 | 21.4 | 19.1 | 16.7 | 19.5 | 21.0 |
| DHFR | 7.7 | 3.9 | 11.5 | 23.1 | 80.8 |
| ER | 9.5 | 17.6 | 17.6 | 13.5 | 28.4 |
| HIV-PR | 13.2 | 17.7 | 19.1 | 14.0 | 16.9 |
| HIV-RT | 2.7 | 2.0 | 4.7 | 4.7 | 2.0 |
| Neuraminidase | 16.7 | 16.7 | 16.7 | 16.7 | 25.0 |
| PTP1B | 12.5 | 12.5 | 12.5 | 12.5 | 50.0 |
| Thrombin | 1.5 | 4.0 | 4.5 | 8.5 | 28.0 |
| TS | 19.4 | 32.3 | 35.5 | 51.7 | 61.3 |
| Average | 11.9 | 15.6 | 17.0 | 20.0 | 33.2 |
| Median | 12.5 | 17.6 | 16.7 | 16.7 | 28.0 |
Table 1: The enrichment factors at 1% screened for various Shape Screening approaches performed according to protocols described by McGaughey et al.7 Increasingly specific atom typing schemes are shown from left to right.
The pharmacophore-based approach also competes very well with other 3D virtual screening methods which have been applied to the McGaughey data set. Table 2 compares Shape Screening pharmacophore-based enrichments to those obtained using the ROCS-color technique9 and the SQW superposition method developed at Merck.7,10 Shape Screening surpasses both of these methods by 30-40% in terms of average and median enrichments, and outperforms each of them head-to-head in eight of 11 cases. Since publication of the McGaughey paper in 2007, ROCS‑color has been viewed by many as the gold standard for shape-based screening, so these latest results are of particular significance.
| Target | EF(1%) | ||
|---|---|---|---|
| Schrödinger Shape Screening | SQW | ROCS-Color | |
| CA | 32.5 | 6.3 | 31.4 |
| CDK2 | 19.5 | 9.1 | 18.2 |
| COX2 | 21.0 | 11.3 | 25.4 |
| DHFR | 80.8 | 46.3 | 38.6 |
| ER | 28,4 | 23.0 | 21.7 |
| HIV-PR | 16.9 | 5.9 | 12.5 |
| HIV-RT | 2.0 | 5.4 | 2.0 |
| Neuraminidase | 25.0 | 25.1 | 92.0 |
| PTP1B | 50.0 | 50.2 | 12.5 |
| Thrombin | 28.0 | 27.1 | 21.1 |
| TS | 61.3 | 48.5 | 6.5 |
| Average | 33.2 | 23.5 | 25.6 |
| Median | 28.0 | 23.0 | 21.1 |
Table 2: A comparison of the Shape Screening pharmacophore-based approach to other 3D virtual screening methods.
Other versatile features of Shape Screening include the ability to score poses in place, force the alignment of specific atoms by way of SMARTS matching, compute similarities to multiple shape queries in a single run, apply alternate similarity normalization schemes that facilitate the identification of embedded shapes, and filter hits using excluded volumes. The Shape Screening technology has also been employed to develop a fast, multi-ligand superposition method, where the template and the structures being aligned to it are all treated in a flexible manner, and the template conformer that yields the best overall alignment of all ligands is utilized.
References
- Kirchmair, J.; Distinto, S.; Markt, P.; Schuster, D.; Spitzer, G. M.; Liedl, K. R.; Wolber, G., How To Optimize Shape-Based Virtual Screening: Choosing the Right Query and Including Chemical Information. J. Chem. Inf. Model. 2009, 49, 678-692.
- Putta, S.; Beroza, P. Shapes of Things: Computer Modeling of Molecular Shape in Drug Discovery. Curr. Top. Med. Chem. 2007, 7, 1514-1524.
- Grant, J.; Pickup, B. A. Gaussian Description of Molecular Shape. J. Phys. Chem. 1995, 99, 3503-3510.
- Rush, T. S., III; Grant, J. A.; Mosyak, L.; Nicholls, A. A Shape-Based 3-D Scaffold Hopping Method and its Application to a Bacterial Protein-Protein Interaction. J. Med. Chem. 2005, 48, 1489-1495.
- Dixon, S.; Smondyrev, A.; Knoll, E.; Rao, S.; Shaw, D.; Friesner, R., PHASE: A New Engine for Pharmacophore Perception, 3D QSAR Model Development, and 3D Database Screening: 1. Methodology and Preliminary Results. J. Comput.-Aided Mol. Des. 2006, 20, 647-671.
- Sastry, M.; Dixon, S. L.; Sherman, W. Rapid Shape-Based Ligand Alignment and Virtual Screening Method Based on Atom/Feature-Pair Similarities and Volume. J. Chem. Inf. Model. 2011, 51(10), 2455-2466.
- McGaughey, G. B.; Sheridan, R. P.; Bayly, C. I.; Culberson, J. C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.-F.; Cornell, W. D., Comparison of Topological, Shape, and Docking Methods in Virtual Screening. J. Chem. Inf. Model. 2007, 47, 1504-1519.
- Sastry, M.; Lowrie, J. F.; Dixon, S. L.; Sherman, W. Large-Scale Systematic Analysis of 2D Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. J. Chem. Inf. Model. 2010, 50, 771-784.
- Hawkins, P. C. D. A Comparison of Structure-Based and Shape-Based Tools for Virtual Screening. Abstracts of Papers, 231st ACS National Meeting, Atlanta, GA, United States, March 26-30, 2006.
- Miller, M. D.; Sheridan, R. P.; Kearsley, S. L. SQ: A Program for Rapidly Producing Pharmacophorically Relevant Molecular Superpositions. J. Med. Chem. 1999, 42, 1505-1514.