JUN 25, 2025
How to find a druggable target: A computational perspective
Roughly 20,000 proteins make up the human proteome, yet not all proteins are suitable drug targets. This makes finding druggable targets a critical early step in drug discovery. Structure-based techniques can enable better, quicker, and more cost effective target tractability assessment. These technologies can also expedite hit discovery and ligand binding potency and selectivity optimization and guide medicinal chemistry rationale.
Join us in this beginner-friendly webinar that will introduce you to strategies and best-in-class tools for identifying druggable, technology-enabled targets. We will review workflows to assess protein binding sites and identify protein-ligand complexes suitable for structure-based drug design. We will also discuss how to reduce uncertainty in structure-based drug models using free energy methods (FEP+), as well as examine how modern physics-based modeling approaches amplified by machine learning can reveal new therapeutic opportunities.
Last, we will highlight a real-world case study from Schrödinger’s Therapeutics Group, showcasing how these strategies were used to assess the druggability of the Wee1 binding site and subsequently how FEP+ was used to enable the discovery of a highly selective Wee1 inhibitor for the treatment of solid tumors. The case study demonstrates the power of computational approaches to unlock challenging targets and drive successful drug discovery programs.
Whether you’re new to modeling or looking to expand your toolkit, this session will provide a practical foundation and a comprehensive guide to help you get started.
Webinar highlights:
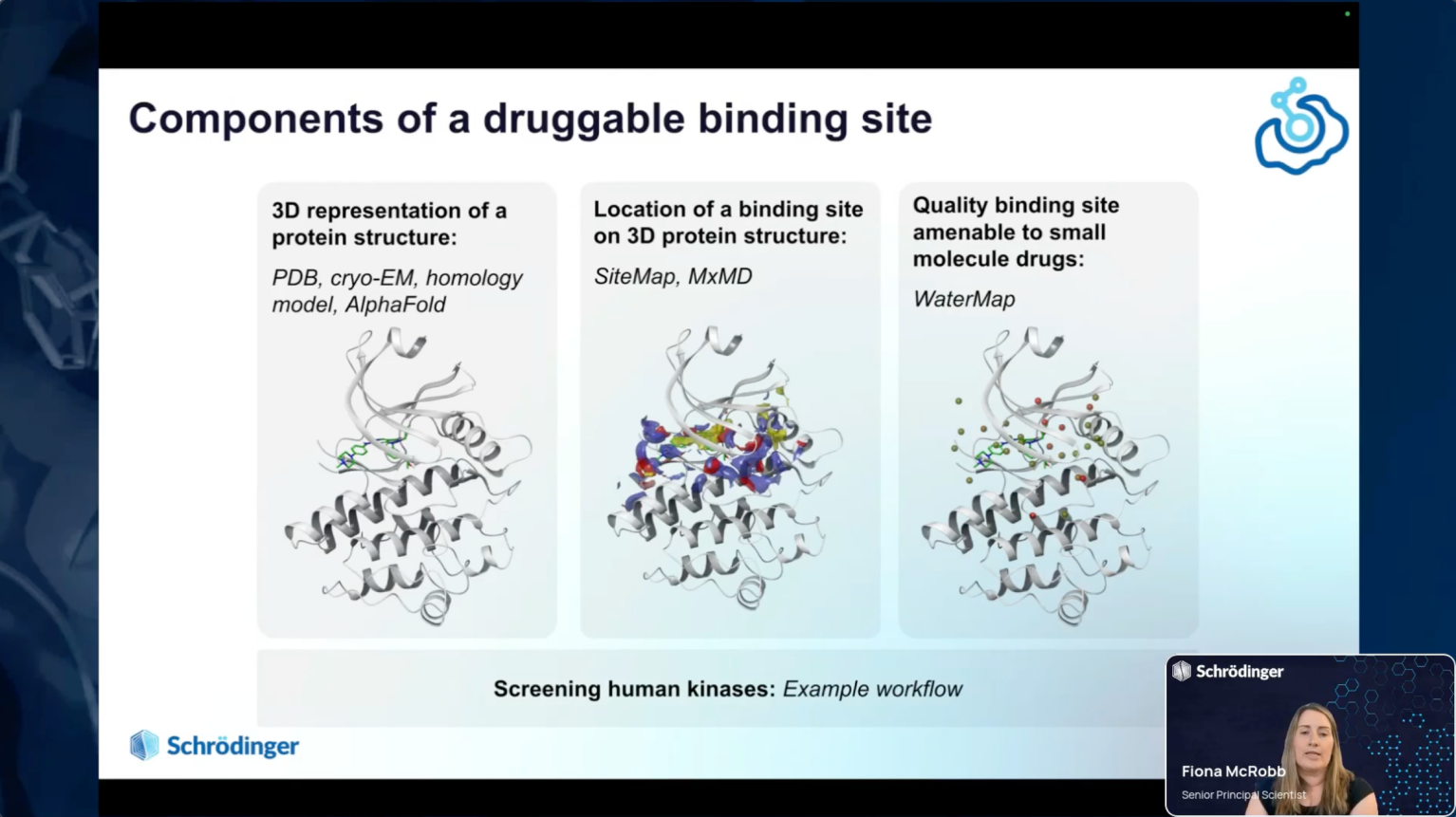
- Discussion of what a computationally druggable target is and how to identify one
- Overview of structure-based techniques, including docking, molecular dynamics, and free energy methods
- Introduction to integrated tools and workflows for binding site detection, druggability assessment, and target prioritization
- Leveraging FEP+ to validate structural models and reduce uncertainty by comparing predicted and experimental ligand affinities
- Case study: Applying these approaches in Schrödinger’s Wee1 inhibitor discovery program
Our Speaker

Fiona McRobb
Senior Principal Scientist, Computational Chemistry, Therapeutics Group, Schrödinger
Fiona McRobb received her Ph.D. in Medicinal Chemistry from Monash University in Australia in 2012. Her Ph.D. encompassed both synthetic and computational approaches to study G protein-coupled receptors (GPCRs). In 2011, she joined Prof. Ruben Abagyan’s lab at UCSD as a postdoc, studying allosteric modulators of GPCRs and environmental toxicology. Fiona joined Schrödinger in 2014 as an Applications Scientist before transitioning to Schrödinger’s Therapeutics Group in 2016. As a Senior Principal Scientist, she applies Schrödinger’s computational chemistry software to address challenging problems in drug discovery, with a focus on identifying new targets and early drug discovery programs.