JUN 12, 2025
Protein and Solvent Dynamics Simulations to Understand Cancer Mutations
Abstract:
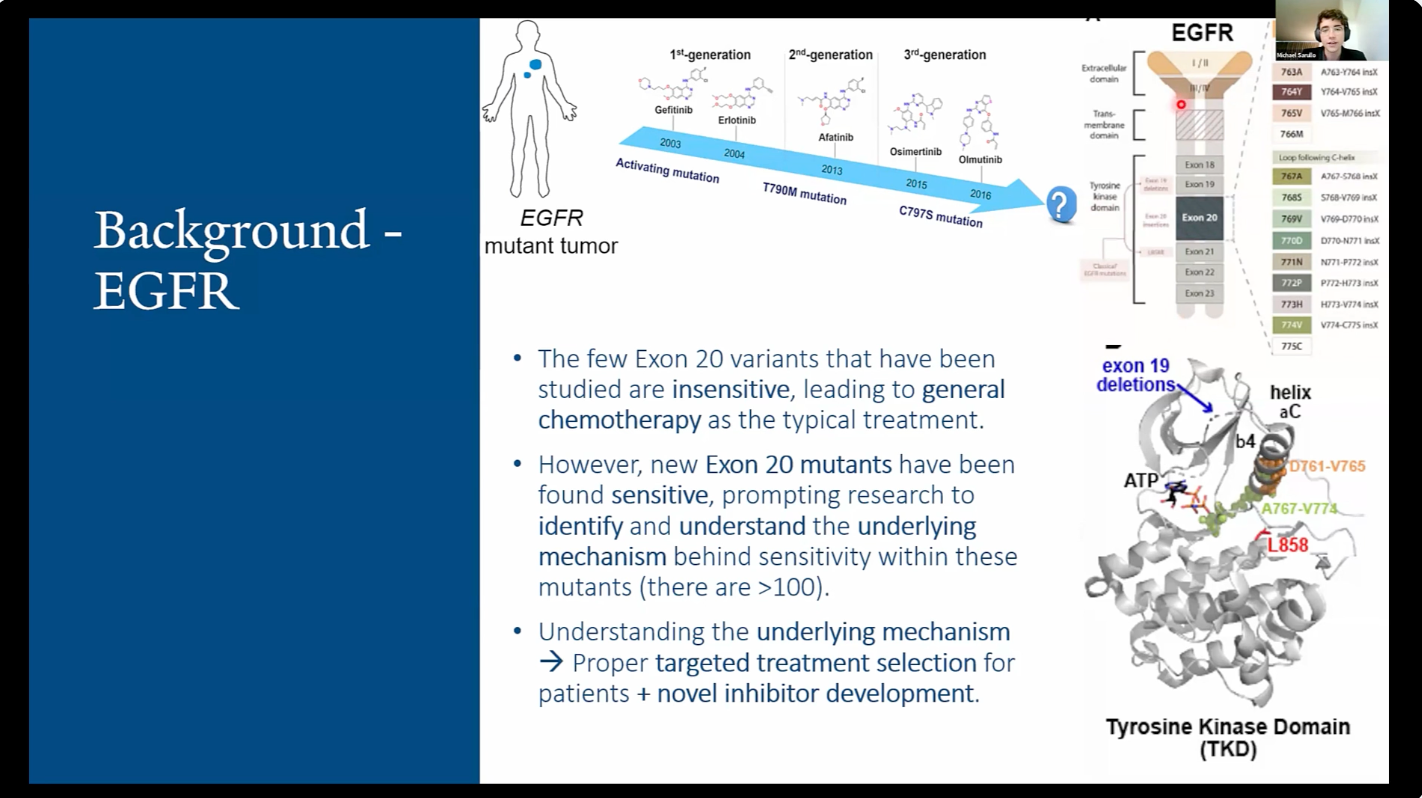
Mutations in a class of protein-based enzymes called kinases cause many cancers, prominently the epidermal growth factor receptor (EGFR) in lung cancer. Drugs that block binding of a key substrate–ATP–can block these cancers, but their effects vary greatly depending on how the kinase is altered by the mutation. However, no clear molecular mechanism has been identified to explain how different mutations affect drug sensitivity. Based on our previous experimental and computational work, we hypothesize that molecular dynamics (MD) simulations paired with various statistical models can classify mutations in terms of drug sensitivity by analyzing coupled protein and solvent dynamics. Using AlphaFold3 to obtain 3D conformations of mutated kinases, I executed MD simulations of known drug-sensitive and drug-resistant EGFR insertions. I constructed a pair-wise dynamical cross-correlation matrix (DCCM) with MDTraj to measure paired secondary-structure fluctuations, fit a logistic regression model to these correlations, and identified features associated with drug sensitivity. My analysis revealed that drug-sensitive mutants display significantly unstable MD trajectories, especially in areas key for ATP binding. Visual analysis of DCCM plots revealed specific clusters of extreme correlations specific to drug-sensitive mutants. Preliminary statistical analysis of correlations revealed ‘breathing’ motions (moving together and apart) between two key parts of the kinase structure in resistant mutations. This association is likely due to a disruption in the integrity of the ATP binding site, allowing drugs to block ATP binding more effectively. Continuing to enhance understanding of the biochemical underpinnings behind drug sensitivity of cancer-causing kinases should lead to streamlined treatment options for lung cancer patients, and deduced structural implications should aid development of future ATP-competitive and allosteric kinase-targeted cancer drugs. Future directions involve analyses of molecular docking experiments paired with principal component analysis (PCA) of trajectories to identify new drug opportunities.
Speaker:
Michael Sarullo, Yale University
Michael Sarullo is a rising third-year undergraduate at Yale University, where he is pursuing a double major in Molecular Biophysics and Biochemistry alongside Statistics and Data Science. His research focuses on computational approaches to understanding biological systems, particularly protein dynamics and their therapeutic applications.