MAY 20, 2020
Automated high-throughput in silico reaction screening for design and discovery of enhanced reactivity and tailored chemo-, regio-, and stereo-selectivity
Speaker:
Thomas Mustard, Principal Scientist
Abstract:
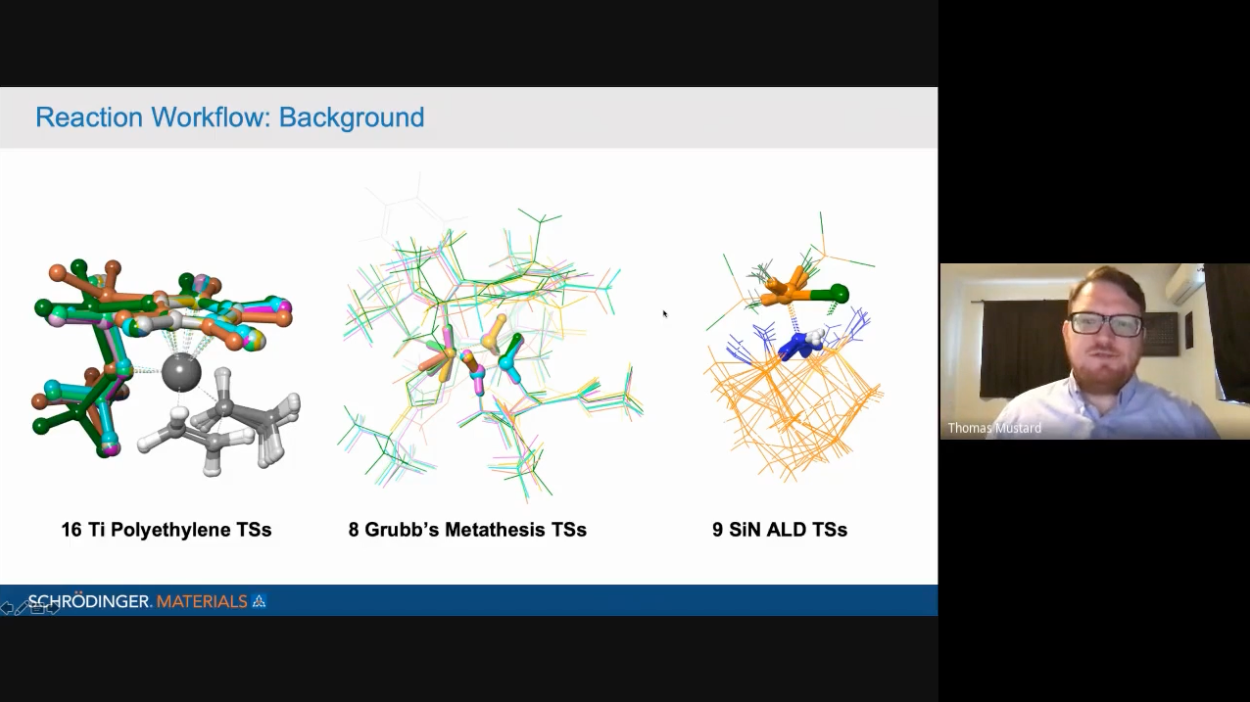
First-principles simulation has become a reliable tool for the prediction of structures, chemical mechanisms, and reaction energetics for the fundamental steps in catalysis. Details of reaction coordinates for competing pathways can be elucidated to provide the fundamental understanding of observed catalytic activity, selectivity, and specificity. Such predictive capability raises the possibility for computational discovery and design of new catalysts with enhanced properties.