SEP 25, 2024
Efficient Computation of Process Parameters for Controlling the Chemistry of Deposition or Etch
Speaker:
Simon Elliott, Research Leader, Schrödinger
Abstract:
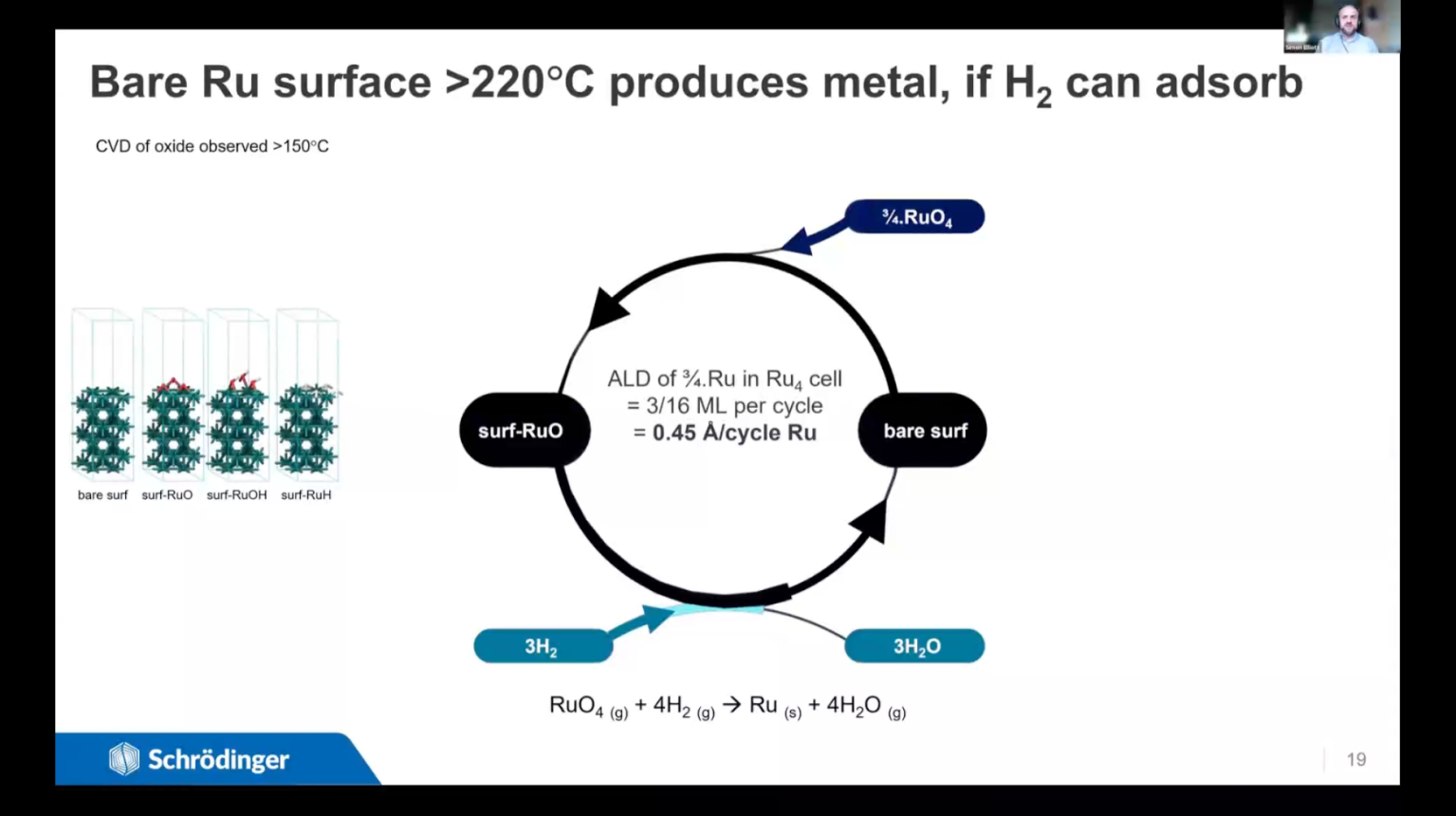
We present a variety of computational techniques for understanding, controlling and improving deposition and etch processes. The emphasis is on choosing the right technique for the research question and time available. The same computational techniques can be used to investigate other gas-surface processes, such as catalysis or sensing. Different chemical processes can be in competition when a solid surface is treated with a gaseous reagent and the outcome is determined by conditions such as temperature and pressure. For instance, continuous deposition (CVD) may take over from self-limiting deposition (ALD) as the temperature is raised. Or temperature may dictate which material is deposited; in the case presented here, ruthenium oxide film is deposited from RuO4+H2 in experiments at 75°C, whereas Ru metal is obtained at 100°C and above. Ru is being investigated as an electroplating seed layer in electronics, as a capacitor electrode and as a heterogeneous catalyst – all applications that require metal rather than oxide. We show that thermodynamics based on density functional theory (DFT) is a computationally-efficient approach for distinguishing between the possible surface-gas processes. The temperatures and pressures for crossover between different chemistries can be estimated, with the accuracy depending on how entropy, coverage and diffusion are treated. We use DFT to examine the conditions of stability for Ru metal, hydride, hydroxide and oxide with respect to H2 and RuO4 reagents, and so explain the crossover from oxide to metal film just below 100°C. We point out how to balance the cost (in terms of researcher time and computer time) against the benefit that each level of accuracy can offer. In the second part of the talk, we introduce Microkinetic Modelling, a new Schrödinger capability for examining the overall kinetics of gas-surface chemistry by solving the coupled kinetic rate equations of its constituent elementary reaction steps. This allows the simulation of macroscopic parameters such as sticking coefficients that can be experimentally measured and used as inputs for fluid dynamics simulations. We first outline the computational scheme, where elementary steps and their activation free energies have been computed with DFT. The resulting microkinetic model for alumina ALD yields measurable quantities (e.g. growth rate) as a function of temperature and pressure, which are validated against experiment. Variation with pressure can account for penetration depth and conformality within high aspect ratio features. The two cases discussed in this talk thus illustrate how atomic-scale DFT can be embedded into higher-level computational schemes for accurate and achievable prediction of the conditions and parameters for controlling chemical processes.