Machine Learning for the Design of Novel OLED Materials
Leveraging Atomic Scale Modeling for Design and Discovery of Next-Generation Battery Materials

SEPT 22, 2022
Leveraging Atomic Scale Modeling for Design and Discovery of Next-Generation Battery Materials
Speaker
Garvit Agarwal
Senior Scientist
Abstract
The development of rechargeable Li-ion batteries (LIBs) has revolutionized electric vehicles and portable electronic devices. Further advancements are needed to improve the power density, safety, reliability, and lifetime of LIBs. Over the past few decades, atomistic modeling of battery materials has complemented experimental characterization techniques and has become an integral part of the development of new technologies. Reliable atomic scale modeling enables rapid initial evaluation of large chemical and material design space accelerating the development cycle of next-generation battery technologies.
In this webinar, we will demonstrate how Schrödinger’s advanced digital chemistry platform can be leveraged to accelerate the design and discovery of next-generation battery materials with improved properties. We will discuss the application of both physics-based and machine learning techniques for understanding structure-property relationships of different components of batteries including electrodes, electrolytes and electrode-electrolyte interfaces. We also discuss the automated active learning framework for the development of state-of-the-art neural network force fields for modeling liquid electrolytes. The framework allows training the force field using highly accurate range-separated hybrid density functional theory data which enables accurate prediction of critical bulk properties of high-performance liquid electrolytes for application in advanced batteries.
Key Learning Objectives:
- Understand predictive capabilities of physics-based modeling for battery materials
- Learn how automated high throughput simulation workflows enable rapid screening of new battery material candidates
- Application of advanced neural network force fields for accurate electrolyte property prediction
Chinese: Driving the development of bio-based polymer materials with molecular simulations | 分子模拟技术推进生物基聚合物材料的发展

SEPT 15, 2022
Chinese Webinar: Driving the development of bio-based polymer materials with molecular simulations | 分子模拟技术推进生物基聚合物材料的发展
Speakers
讲师:Miao Shi, Ph.D., Schrödinger, Inc
嘉宾:Lihua Chen, Ph.D., Schrödinger, Inc
Summary
生物基聚合物(由可再生资源制成的聚合物材料)的应用正在为各个行业,从消费品包装到碳纤维复合材料,带来整体效益。这项材料的转变对研发和制造团队的现有经验提出了挑战,团队需考虑如何从石油基聚合物扩展到这些新系统。分子模拟为团队了解生物基聚合物的行为方式以及如何有效地在配方中使用它们提供了一个关键窗口。
本次网络研讨会将为研发管理者、材料科学家和工程师以及聚合物科学家提供学习聚合物模拟技术的机会以及筛选和评估生物基聚合物材料性能的案例。
通过参加本次网络研讨会,您将了解:
- 生物基材料开发的新数字方法
- 分子模拟如何缩短配方开发周期
- 确定您研发过程中分子模拟可提供价值的关键领域
Accelerating Digital Drug Design With Automated Informatics Workflow


SEPT 8, 2022
Accelerating Digital Drug Design With Automated Informatics Workflow
Speakers
Jay McGill, PhD, Chief Operating Officer, IBRI
Mary Mader, PhD, VP Molecular Innovation, IBRI
Vipin Vijayakumar, VP Research Infrastructure, Maze Therapeutics
Abstract
In this webinar, research leaders from Maze Therapeutics and Indiana Biosciences Research Institute (IBRI) will discuss their best practices in managing and deriving insights from complex data by leveraging best-in-class informatics tools to create discovery workflows that span data capture and organization to compound design and analysis.
Learning Objectives:
- How do informatics technologies drive scientific discoveries
- What are the biggest data challenges to new companies and what are the recommended ways to address them
- How to select the right tools based on the problems to be addressed
Exploring the formulations of personal care products using a digital chemistry strategy


AUG 30, 2022
Exploring the formulations of personal care products using a digital chemistry strategy
Speaker
Jeffrey Sanders
Product Manager of Consumer Packaged Goods
Abstract
The demand for new and innovative personal care products is increasing due to changes in consumer trends and sustainability goals for CPG companies. Customers are becoming more discerning – choosing products based on understanding the ingredients and whether a product is made with natural or petroleum-based materials. These concerns highlight challenges for consumer goods research and development. To meet these challenges and retain their position in the consumer marketplace, new product formulations need to be developed and match the existing formulation’s key properties. Understanding how ingredients behave in products and “in action” will be necessary to drive not only new development but also end-to-end product tracing. To streamline this process, multi-scale physics simulations can be utilized to cut down product development timeline and cost, as well as optimize large-scale production by simulating digital twins. Molecular simulation provides a unique opportunity to predict how individual ingredients will behave in formulations. Atomistic simulations can help researchers and engineers understand product morphology, solubility, and other physical properties if the components are known. Unlike process simulations, only the chemistry and composition are required to build molecular models of up to millions of atoms and predict properties. Beyond physics-based modeling, chemical information can be used to build machine-learned models with existing experimental or sensory data. In this talk, we will show you molecular modeling in action and explore how digital chemistry strategies are driving innovation in personal care product formulations.
Learning Objectives:
- How to gain insight of individual ingredient behavior and key properties of components in formulation using multi-scale physics simulations
- How to predict key properties of formulations with advanced machine learning
- How digital chemistry can accelerate your research and development in personal care product
Battery and energy storage materials
Battery and energy storage materials
Background
The design and manufacturing of safer, less expensive, and more effective energy storage devices is a critical challenge in a wide variety of industries including the automotive, aviation, and energy sectors with societal and environmental implications. Atomic-scale materials modeling has become an essential tool for the development of novel battery components — cathodes, anodes, and electrolytes — that support higher power density, capacity, rate capability, faster charging, and improved degradation resilience. Schrödinger’s Materials Science software platform provides a powerful atomic-scale modeling solution for comprehensive analysis of ion diffusion, mechanical response, and electrochemical response in electrodes and electrolytes, dielectric properties of potential electrolyte compounds, and other relevant properties.
Application: Virtual screening for sei forming li-ion battery additives
Additives in electrolyte formulations are designed to increase dielectric strength and electrode stability by facilitating the formation of a stable solid electrolyte interphase (SEI). To enhance stability of the SEI, these electrolyte additives should exhibit high reduction potential and increased reactivity. From a molecular design perspective, the former is related to the energy of the lowest-unoccupied molecular-orbital (LUMO), while the latter is correlated with molecular hardness. Using these two key molecular descriptors, one can screen through a large library of compounds to find the ideal candidates for new electrolyte additives. Figure 1 illustrates the final stage of a virtual screening example applied to an automatically generated library of 368,073 unique electrolyte compounds with 6 cyclic carbonate and sulfone cores and 18 R-groups. The final list of compounds showcases a chemical design space pregressively screened using the accurate force field (OPLS2005 [1]), and semiempirical quantum chemistry (RM1 [2]), followed by a final selection process based on a full quantum chemical analysis with density functional theory (DFT).
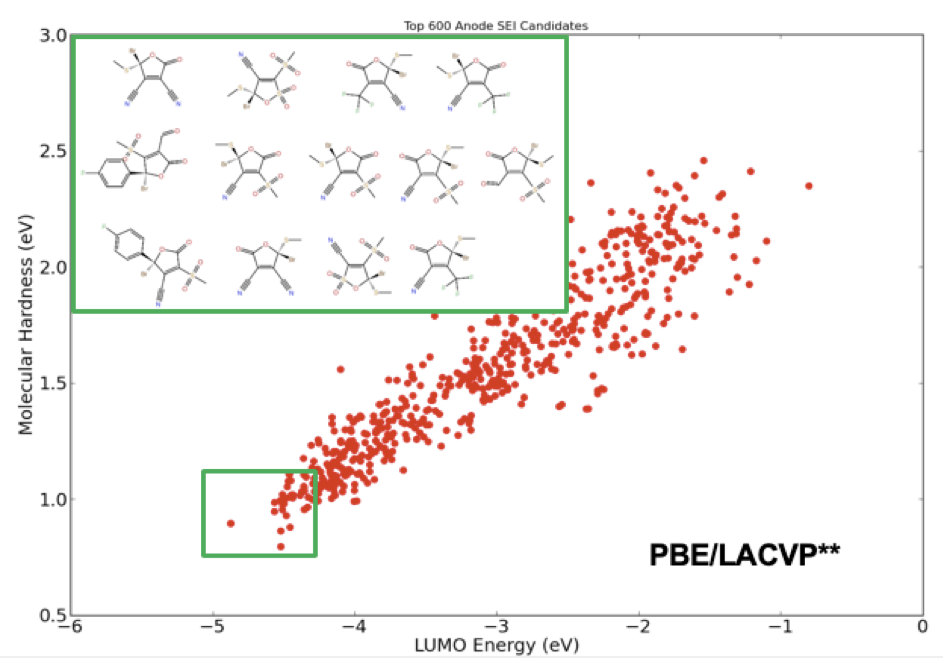
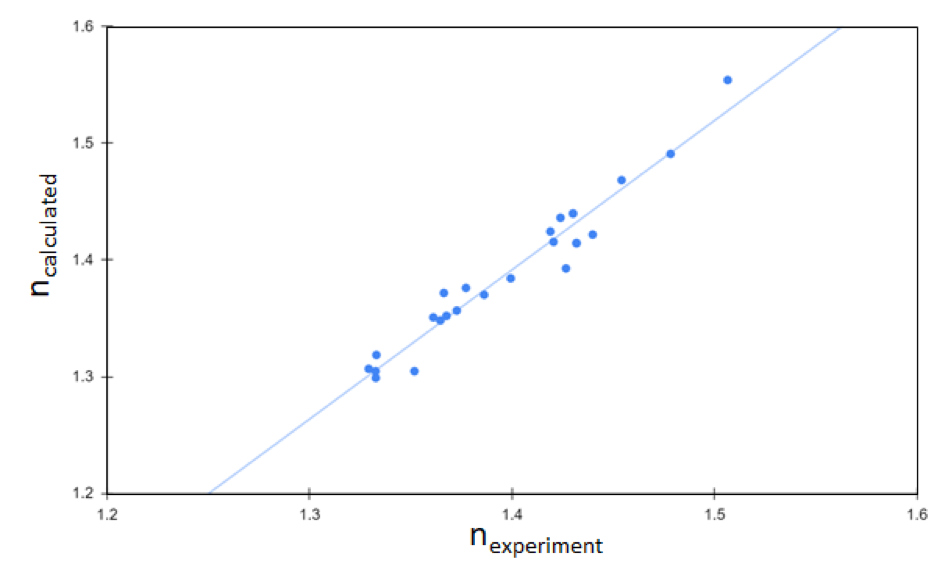
Application: Dielectric properties of molecular electrolytes
The dielectric constant is another key design factor for battery electrolytes. Since the dielectric constant correlates directly with optical response for compounds with relatively low polarity, screening for compounds with high refractive index using the Schrödinger Materials Science Suite can quickly narrow down the list of potential molecular electrolytes to those with desirable dielectric properties. A study summarized in Fig. 2 showcases the accuracy of computational prediction for refractive indices of molecular liquids. As shown in Fig. 2, the calculated dielectric constants agree well with experimentally measured values [5], validating the accuracy of theoretical predictions. This predictive capability is a direct result of (1) the speed and accuracy of compute engines used by Schrödinger software and (2) the seamless integration between two independent simulation methods – quantum mechanics (QM) and classical molecular dynamics (MD). This example shows how theoretical predictions at the atomic-scale assess key intrinsic properties of battery materials, such as dielectric properties.
Application: Intercalation energetics of electrode materials
Ion intercalation in electrodes is a critical process that determines the core performance of rechargeable batteries. The Schrödinger Materials Science platform’s periodic DFT engine, Quantum ESPRESSO, has proven its scientific value over a wide range of applications, including the analysis of thermodynamics and kinetics of ion intercalation processes in electrodes.
Work by Arroyo-de Dompablo and coworkers [3] apply Quantum ESPRESSO to design and characterize LiMXO4 cathode materials based on computationally predicted intercalation voltages [3]. Schrödinger’s comprehensive list of modeling and simulation workflow solutions coupled with Quantum ESPRESSO helps to extend these types of analysis to cover broad chemical space and properties to facilitate the research of novel electrode materials.
Apart from intercalation potentials, there is a number of other electrode properties that can be studied with computational methods. Some important examples include ion migration kinetics and pathways, mechanical properties, the volume change upon ion intercalation, electronic properties, chemical stability, and reactivity with respect to electrolyte.
Application: Analysis of polymer electrolyte using classical molecular dynamics
Faster diffusion of lithium ions in battery electrolytes is desirable to improve battery performance. In a recent publication [4], Brooks and coworkers performed thorough investigations of lithium ion diffusion in polyethylene oxide (PEO) electrolyte. Using the Schrödinger Materials Science platform and its classical MD workflows, the authors studied the key diffusion mechanisms of lithium ions in a PEO electrolyte mixed with bistriflimide (also known as TFSI) anions. The lithium ion diffusion coefficient was examined with varying temperature and Li:TFSI ratio [4]. Analysis of the mean square displacement diffusion rates and Li/polymer coordination suggest that PEO is too flexible causing it to bind the lithium ions too tightly, which implies that a less flexible polymer blend used for electrolyte could lead to a faster lithium ion diffusion.
Apart from the ion diffusion itself, the details of coordination between the ion and the electrolyte can be captured and compared, helping to guide research to identify the best chemical groups. Thermophysical and mechanical properties of the polymer electrolytes are also accessible through molecular dynamics simulations expanding the performance criteria of solid electrolytes that can be considered in early screening.
Summary
Atomic-scale simulation and modeling technologies integrated within Schrödinger’s Materials Science software provide critical insight in all facets of the materials design process for battery components- electrolytes, electrodes, and formation of stable SEI. Schrödinger’s comprehensive list of solutions can elucidate key chemical processes of the materials and characterize their crucial thermophysical properties, which can boost the cost effectiveness of the design pipeline for novel materials and accelerate the development of novel energy storage devices.
Selected publications
-
Banks, Jay L., et al. “Integrated modeling program, applied chemical theory (IMPACT).”
Journal of computational chemistry 26.16 (2005): 1752-1780.
-
Rocha, Gerd B., et al. “RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I.”
Journal of computational chemistry 27.10 (2006): 1101-1111.
-
Arroyo-de Dompablo, M. E., et al. “On-demand design of polyoxianionic cathode materials based on electronegativity correlations: An exploration of the Li2MSiO4 system (M= Fe, Mn, Co, Ni).”
Electrochemistry Communications 8.8 (2006): 1292-1298.
-
Brooks, Daniel J., et al. “Atomistic Description of Ionic Diffusion in PEO–LiTFSI: Effect of Temperature, Molecular Weight, and Ionic Concentration.”
Macromolecules 51.21 (2018): 8987-8995.
-
CRC Handbook (85th Ed); Kim, Sunghwan et al. “PubChem 2019 update: improved access to chemical data.” Nucleic acids research vol. 47,D1 (2019): D1102-D1109. ; Gregory, Andrew, R. N. Clarke.
“Tables of the complex permittivity of dielectric reference liquids at frequencies up to 5 GHz.” National Physics Laboratory MAT 23 (2011).
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
ePharmaLib: A Versatile Library of e-Pharmacophores to Address Small-Molecule (Poly-)Pharmacology
生物制药设计 | BioLuminate

JUN 30, 2022
生物制药设计 | BioLuminate
Speaker
Jianxin Duan
Fellow
Abstract
本培训我们将演示BioLuminate生物制药设计工作流程,其中包括:
- 抗体模拟,包括建模和人源化流程
- 蛋白突变 – 亲和力和稳定性预测
- 蛋白表面分析和蛋白聚集性预测
- 序列对比和同源建模
如果需要了解这次或其他的讲座请点击资讯动态网页。
讲师:段践辛博士,薛定谔公司
时间:北京时间下午3点
日期:6月30日,星期四
Molecular Modelling to Support Drug Formulation for Small Molecule and Biologic Drugs

JUN 28, 2022
Molecular Modelling to Support Drug Formulation for Small Molecule and Biologic Drugs
Speaker
John C. Shelley
Fellow
Abstract
- Complementary use of machine learning and physics-based modeling contribute to the drug development and formulation process
- Molecular modelling provides a basic understanding of the structure and behaviour of drugs as formulated that compliments experimental data and informs decision making in drug formulation
- API and excipient physical and chemical property prediction for small molecule drug formulations
- Characterization of drug-drug and drug-excipient association including drug-polymer interactions in small molecule and biologics formulations
- Provide structural insights into concentrated protein solutions and predict viscosity, aggregation, and the effect of excipients
A paradigm change in the design and optimization of OLED materials using a digital chemistry strategy

JUN 22, 2022
A paradigm change in the design and optimization of OLED materials using a digital chemistry strategy
Speaker
Hadi Abroshan
Senior Scientist
Abstract
Recent developments in device architecture of organic light emitting diodes (OLEDs) have opened a new avenue for innovative technologies to fabricate ultra-thin, flexible, foldable, and transparent displays. However, commercial advancement of OLEDs with higher performance requires continued discovery and development of novel optoelectronic materials. Given the enormous chemical design space available, traditional approaches based on chemical intuition and trial-and-error experimentation are expensive, time-consuming, and most often ineffective. A paradigm change in materials design and development is required to realize next-generation OLEDs.
In this webinar, we will present the impact of in silico technologies for systematic design, development, and selection of organic optoelectronic materials. Both physics- and machine learning-based approaches (e.g. active learning, goal-directed generative model) will be discussed. These computational approaches enable development of a better understanding of structure-function relationships from a molecular and morphological perspective. We also demonstrate accelerated OLED materials analysis through the combination of atomic-scale simulations with machine learning to pre-screen novel materials for high performance before laborious synthesis and device fabrication.
Key Topics Covered:
- Understand the predictive capabilities of physics-based modeling in optoelectronics
- Explore new machine learning capabilities for high-throughput screening to accelerate OLED materials discovery