Protein Preparation Workflow
An easy-to-use tool for correcting common structural problems and creating reliable, all-atom protein models
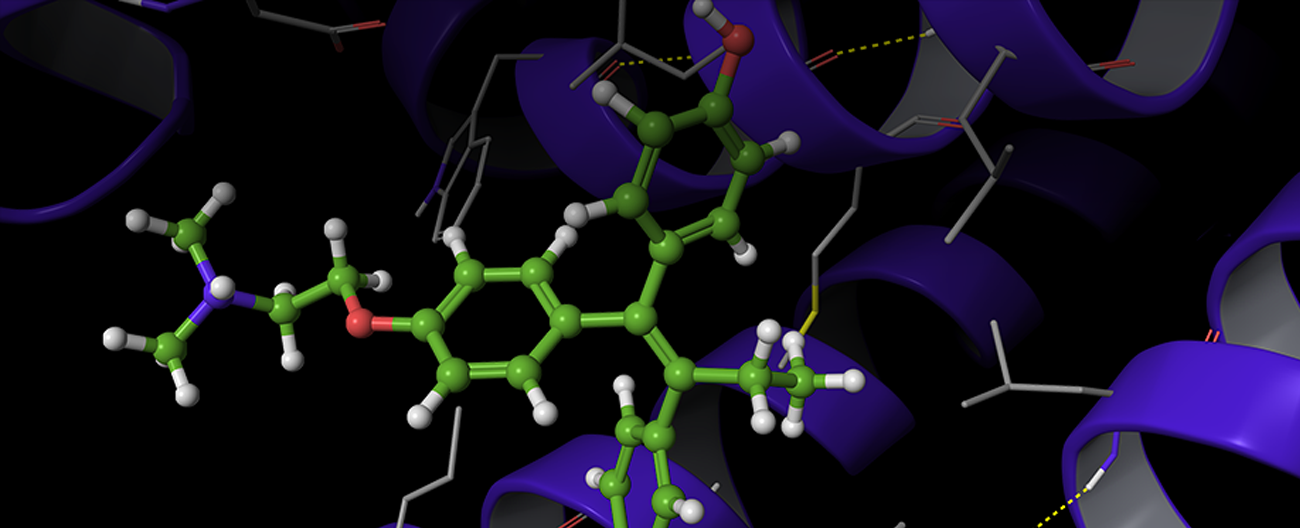
Overview
Successful structure-based modeling projects demand not only accurate software, but accurate starting structures as well. Left untreated, common problems with experimentally-derived structures can lead to wasted time and resources. Schrödinger’s Protein Preparation Workflow is designed to help researchers ensure structural correctness at the outset of a project, equipping them with a high-confidence structure ideal for use with a wide variety of modeling applications.
Experienced modelers know that accurate starting structures are a prerequisite for successful computational drug design. Unfortunately, even when working with a high-resolution x-ray crystallographic structure, researchers can spend considerable time and effort correcting common problems such as missing hydrogen atoms, incomplete side chains and loops, ambiguous protonation states, and flipped residues.
The Protein Preparation Workflow aggregates, automates, and integrates the most frequently used tools and techniques in structure preparation, without shoehorning the researcher into a single inflexible process. Throughout the preparation workflow, a user can choose whether or not to apply any given operation, and because intermediate structures are all organized in the project table, it becomes trivial to share any result with a colleague or use outside applications when a specialized approach may be called for.
More than just a handful of utilities for minor structural corrections, the Protein Preparation Workflow is a robust solution for ensuring a reasonable starting point at the outset of structure-based drug design projects, making it an attractive tool of choice for any chemist whose work relies upon accurate protein models.
Features
Performance
Using Schrödinger’s Protein Preparation Workflow, researchers can convert a raw PDB structure into all-atom, fully prepared protein models in minutes instead of hours or days, while also ensuring the accuracy of all downstream modeling simulations.
The Protein Preparation Workflow enables this increased efficiency in structure preparation by including tools which allow you to:
- Automatically import full PDB files — or any chain within a PDB file — from local databases or the PDB website
- Automatically add missing hydrogen atoms
- Correct metal ionization states to ensure proper formal charge and force field treatment
- Enumerate bond orders to HET groups
- Remove co-crystallized water molecules at the user’s discretion
- Cap protein termini with ACE and NMA residues
- Highlight residues with missing atoms or multiple occupancies
- Pre-process structures for Prime, Schrödinger’s program for protein structure prediction
- Easily navigate between different residues, HET groups, and chains using intuitive graphical tools
- Quickly and easily determine the most likely ligand protonation state as well as the energy penalties associated with alternate protonation states
- Determine optimal protonation states for histidine residues
- Correct potentially transposed heavy atoms in arginine, glutamine, and histidine side chains
- Optimize the protein’s hydrogen bond network by means of a systematic, cluster-based approach, which greatly decreases preparation times
- Perform a restrained minimization that allows hydrogen atoms to be freely minimized, while allowing for sufficient heavy-atom movement to relax strained bonds, angles, and clashes
References
-
Disulfide Bond Engineering of an Endoglucanase from Penicillium verruculosum to Improve Its Thermostability
-
A novel method for in silico assessment of Methionine oxidation risk in monoclonal antibodies: Improvement over the 2-shell model
-
Novel, Self-Assembling Dimeric Inhibitors of Human β Tryptase
-
Clobetasol Propionate Is a Heme-Mediated Selective Inhibitor of Human Cytochrome P450 3A5
-
Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia
-
Predicting mutations deleterious to function in beta-lactamase TEM1 using MM-GBSA
-
Characterizing the Chemical Space of ERK2 Kinase Inhibitors Using Descriptors Computed from Molecular Dynamics Trajectories
-
Adverse Drug Reactions Triggered by the Common HLA-B*57:01 Variant: A Molecular Docking Study
-
Predicting the Effect of Amino Acid Single-Point Mutations on Protein Stability—Large-Scale Validation of MD-Based Relative Free Energy Calculations
-
Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation
-
Selection of Nanobodies that Block the Enzymatic and Cytotoxic Activities of the Binary Clostridium Difficile Toxin CDT
-
Accurate Binding Free Energy Predictions in Fragment Optimization
-
The marine-derived sipholenol A-4-O-3′,4′-dichlorobenzoate inhibits breast cancer growth and motility in vitro and in vivo through the suppression of Brk and FAK signaling
-
Discovery of Thienoquinolone Derivatives as Selective and ATP Non-Competitive CDK5/p25 Inhibitors by Structure-Based Virtual Screening
-
Mechanistic and Computational Studies of the Reductive Half-Reaction of Tyrosine to Phenylalanine Active Site Variants of d-Arginine Dehydrogenase
-
Physics-Based Enzyme Design: Predicting Binding Affinity and Catalytic Activity
-
A Computational Approach to Enzyme Design: Predicting ω-Aminotransferase Catalytic Activity Using Docking and MM-GBSA Scoring
-
Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments
-
Improved docking of polypeptides with Glide
-
Boosting virtual screening enrichments with data fusion: Coalescing hits from two-dimensional fingerprints, shape, and docking