WaterMap
State-of-the-art, structure-based method for assessing the energetics of water-solvating ligand binding sites for ligand optimization

State-of-the-art, structure-based method for assessing the energetics of water-solvating ligand binding sites for ligand optimization

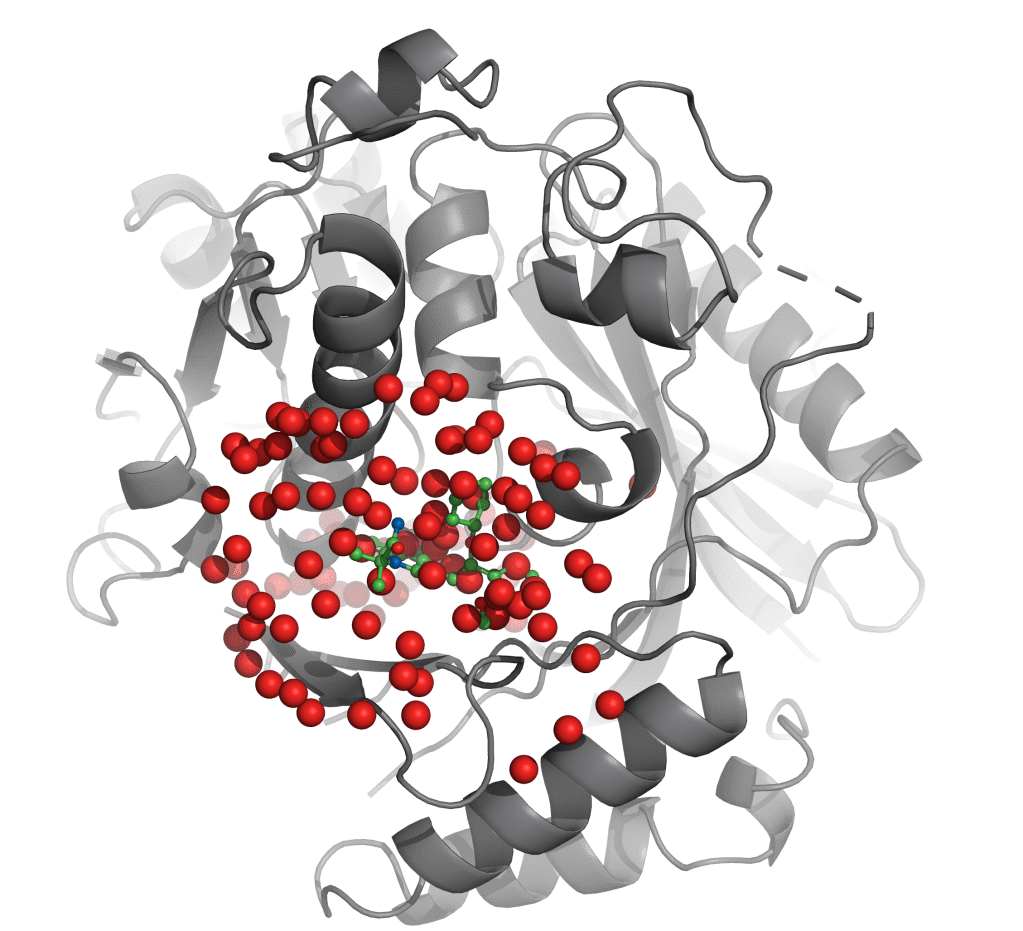
WaterMap is an advanced solution for the reliable calculation of positions and energies of solvating water in a protein binding pocket. By providing key insights to guide ligand design and optimization, WaterMap is a high impact solution for structure-based drug discovery, as demonstrated in several successful drug discovery programs now in clinical development.

See how Nimbus Therapeutics identified potent, selective inhibitors using a virtual screening workflow guided by hydration energetics.
read the case studyBrowse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Get answers to common questions and learn best practices for using Schrödinger’s software.
Learn more about the related computational technologies available to progress your research projects.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.