Accurate and rapid conformation generation with ConfGen
Performance in Reproducing Bioactive Ligand Conformations
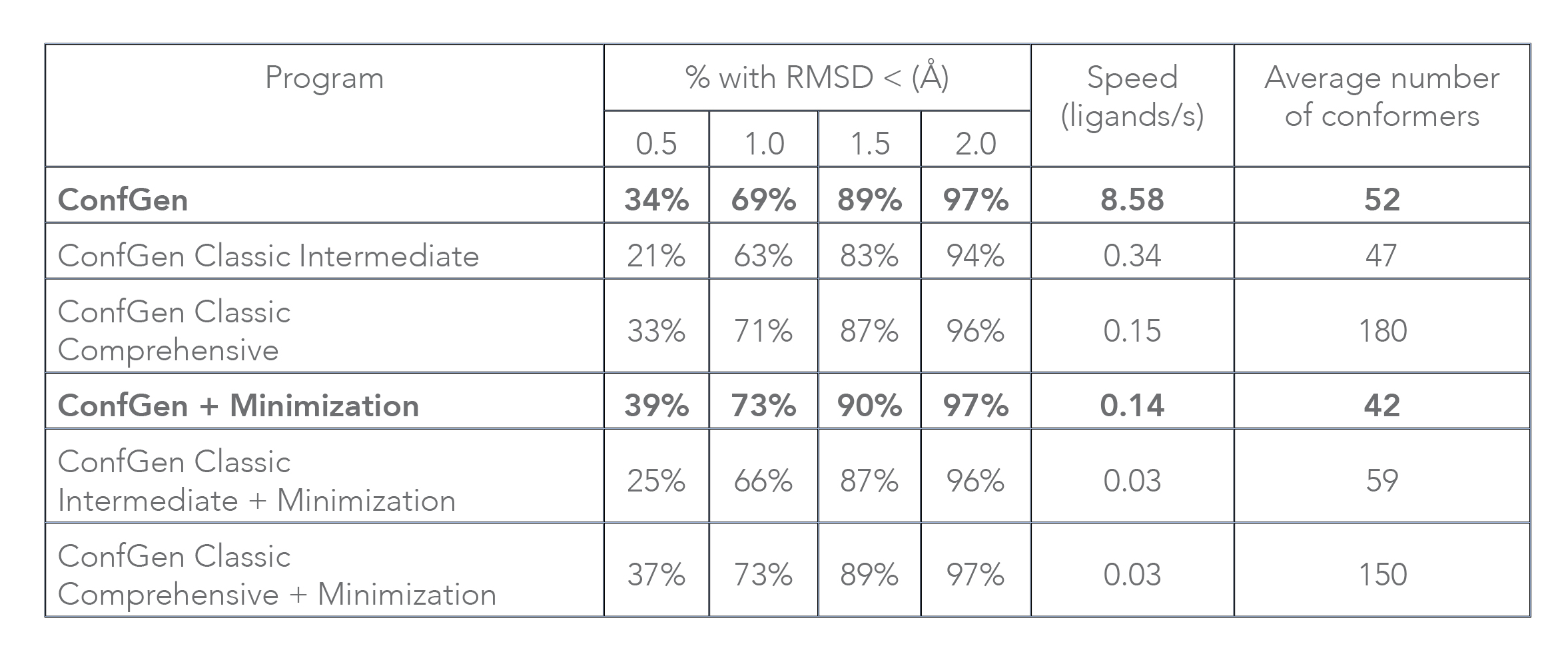
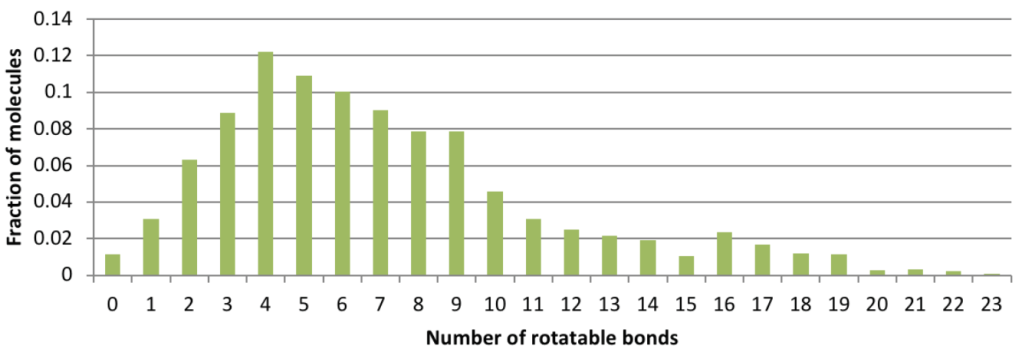
Conformation generation, a procedure for producing diverse, low-energy 3D structures of compounds, is an essential element of in silico drug design, heavily employed in ligand-based virtual screening. To improve the recovery of bioactive conformations among generated conformers and drastically speed conformer generation, Schrödinger has completely rewritten the ConfGen application. As shown in Table 1, the new algorithm is approximately one order of magnitude faster than the previous version of ConfGen (henceforth referred to as ConfGen Classic), with bioactive recovery equivalent to the most computationally intensive mode of ConfGen previously available. This case study utilized 1,904 ligands, extracted from high quality protein-ligand crystal structures. This dataset includes a broad diversity of functional groups present in drug-like molecules and a range of molecule sizes and flexibility, as shown in Figure 1.
Without minimization of output conformers, ConfGen finds the bioactive conformation with an RMSD of < 1.5 Å in 89% of cases compared to 83% and 87% with ConfGen Classic Intermediate and Comprehensive modes, respectively. The new algorithm processes ligands 25 and 57 times faster than ConfGen Classic Intermediate and Comprehensive modes. When minimization of conformers is performed, ConfGen recovers more bioactive conformations with < 1.0 Å RMSD than without minimization. Because minimization acts as the speed bottleneck, the new algorithm is 5 times faster than both ConfGen Classic Intermediate and Comprehensive modes. It should be emphasized that ConfGen is able to achieve bioactive recovery similar to ConfGen Classic Comprehensive in far fewer conformers.
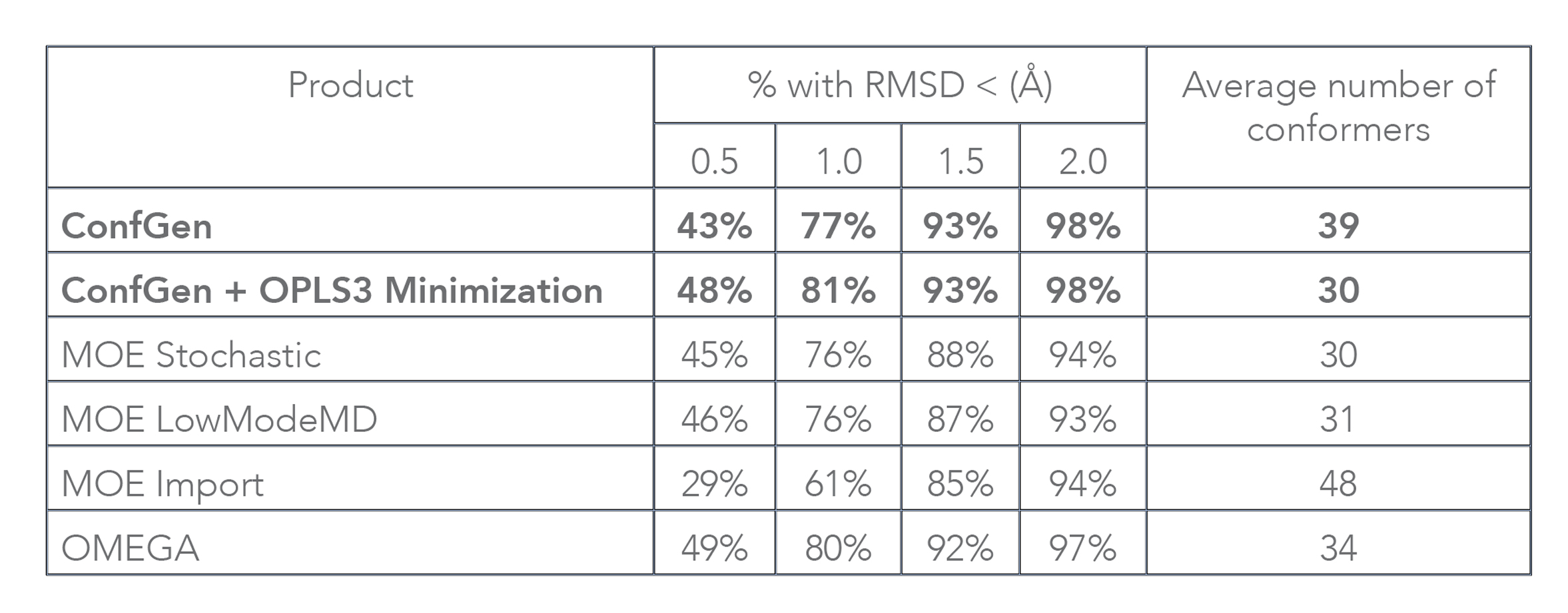
Both ConfGen and ConfGen Classic, along with several other commercial conformer generators, have been benchmarked at the Universität Hamburg [1] on a high quality dataset of 2,859 protein-bound ligand conformations extracted from the PDB. In terms of accuracy, the ConfGen program was found to be on par with OMEGA and overall better than MOE (see Table 2). Note that this paper assessed processing time by running conformer generator programs for a single ligand at a time, which is not representative of typical use. In addition, overhead from Schrödinger’s job-control system that enables easy submission to remote queues and hosts was included as part of the ConfGen timing. With these contributions factored out, when applied to the dataset used in that study, ConfGen (Suite 2017-3 release) processes on average 15 ligands per second without optimization, and about 0.4 ligands per second with geometry optimization using OPLS3. These speeds are similar to OMEGA’s and at least an order of magnitude faster than MOE Import, the fastest MOE approach.
ConfGen Algorithm
The new ConfGen algorithm utilizes a divide-and-conquer strategy to build feasible molecular conformations in three steps: (1) divide input molecules into fragments by breaking exo-cyclic rotatable bonds; (2) obtain conformations for all the fragments; (3) build conformations for the whole molecule by reconnecting the fragments in various ways.
The following types of bonds are considered non-breakable for fragmentation purposes: ring bonds, double or triple bonds, bonds to terminal atoms, and common types of “rigid” bonds such as N-C bonds in amides and thioamides and O-C bonds in esters and thioesters. All other bonds are broken and capped by hydrogens during fragmentation. This process is illustrated in Figure 2.

An examination of the Zinc database [2] was used to identify approximately 40,000 template fragments. A MacroModel conformational search was applied to each of these fragments to generate a library of conformations for use in ConfGen searches. Conformations for fragments not found in the library are generated on the fly via a two-stage optimization process starting from random initial atom positions.
The first optimization stage uses a penalty function to efficiently obtain crude coordinates while the second stage uses the OPLS3 force field to fine-tune the conformations. The process is repeated a number of times depending on the number of sp3 centers in the fragment. By studying compounds from several public databases, it was observed that at most roughly 2n⁄2 low-energy conformations, where n is the number of sp3 centers, are expected for the fragments generated using the ConfGen fragmentation rules. The algorithm generates this many conformations up to a maximum of 24 by default. Conformations with heavy atom RMSD values less than a configurable threshold (0.25 Å by default) relative to previously generated conformations are classified as duplicates and discarded.
The penalty function used in the first step of the on-the-fly conformer generation for fragments consists of terms designed to produce the desired features of the conformations:
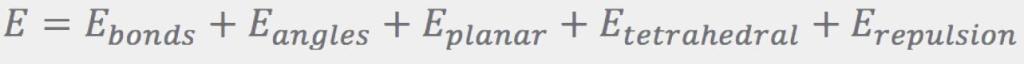
The Ebonds contribution is a sum of harmonic terms (one for every bond) that penalize departures of the bond lengths from their ideal values:
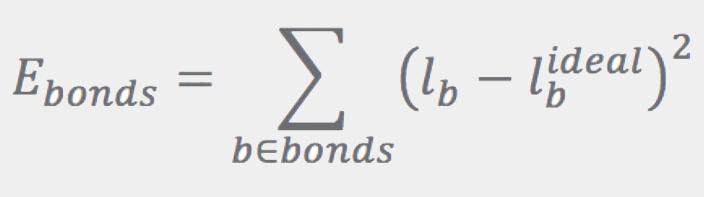
Ideal bond lengths, lbideal, are obtained as described in Refs. [3] and [4]. The Eangles term has the same form as Ebonds, except that the sum runs over angles. Ideal angles are determined using simple rules based on atom hybridization, number of connections, and ring membership. Instead of using the angles directly, Eangles uses the distance between the first and third atoms in the angle, la:
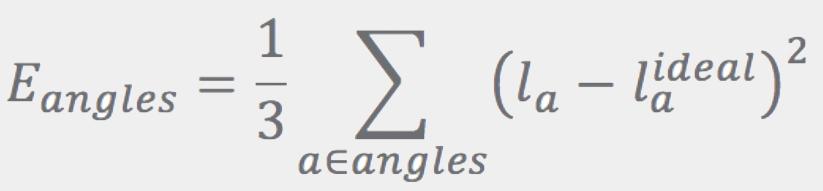
Both of the Eplanar and Etetrahedral terms are given by the sums over “templates”:
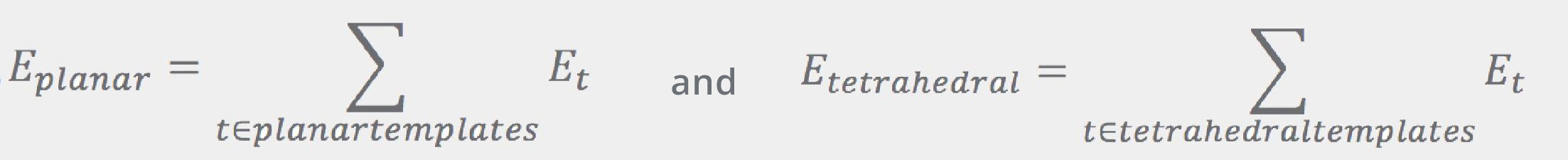
where Et is proportional to the RMSD of the fragment’s atoms with regard to the corresponding atoms in the template:
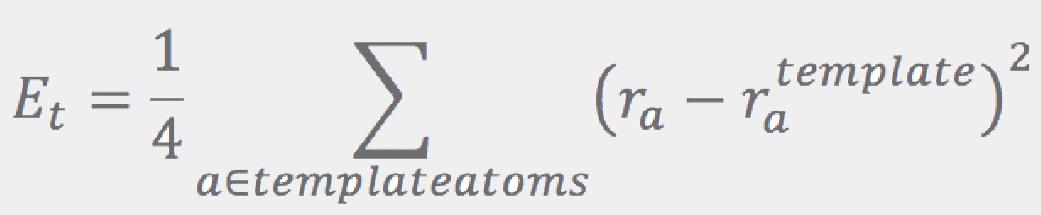
Here, template atoms are assumed to be superimposed onto the corresponding fragment’s atoms. ConfGen uses three kinds of templates: one for tetrahedral centers (3 or 4 atoms around an sp3 center) and another two for the planar configurations around sp2 atoms and double bonds. In all cases the ideal (template) positions are built using ideal bond lengths and “perfect” angles (120° for the planar templates and 109.5° for the tetrahedral). Finally, the Erepulsion term is given by a sum of short-ranged soft “bumps” over the non-bonded atom pairs (that is, pairs of atoms that do not contribute to Ebonds or Eangles):
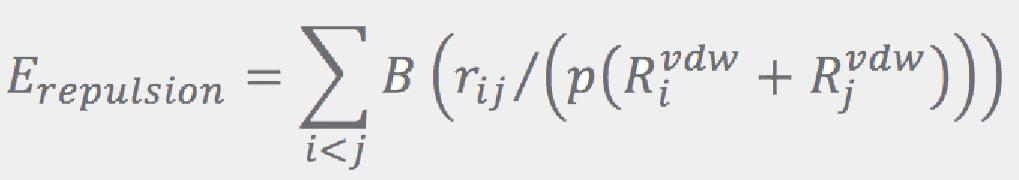
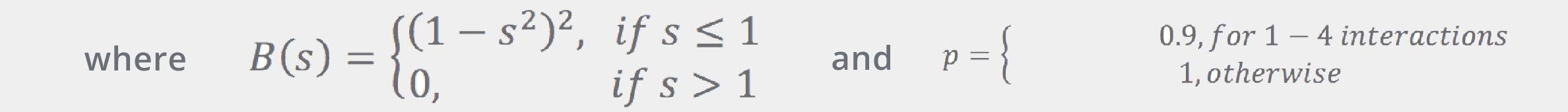
where rij are the interatomic distances, and RivdW are the van der Waals radii (from Ref.[5]).
The fragments are reconnected along bonds broken in the fragmentation step, one bond at a time. The linking order is chosen to favor joining similarly sized portions of the molecule at each intermediate step. Specifically, at each level, conformations for two fragments (A and B) are used to create the conformations for the combined fragment A+B. First, SMARTS patterns matched against the whole molecule are used to select specific dihedral angles to sample along the A − B bond. When there is only one conformation available for either (or both) of the fragments, the fragment’s symmetry is first identified by rotating it along the new bond by 45, 60, 90, and 180 degrees to see if equivalent structures result.
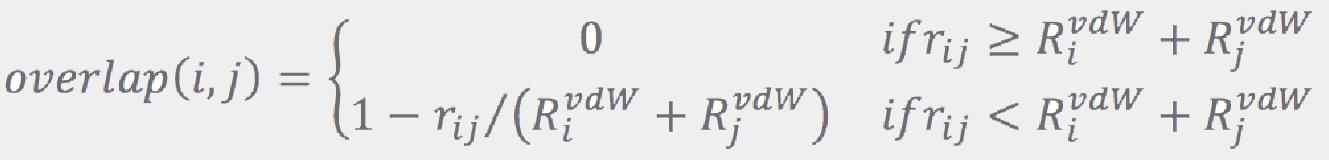
where rij is the distance between atoms i and j, and RijvdW are their van der Walls radii from Ref. [5]. If the largest overlap is less than a configurable threshold (0.5 Å by default), the combination is accepted as a viable candidate. This process is repeated until a prescribed number of candidates (96 by default) is collected. If no combination passes the overlap test, all of them are considered to be viable candidates. A prescribed number of the best ranked candidates is then retained (64 by default), while the others are dropped. More conformations are retained for larger molecules: if the number of rotatable bonds exceeds some threshold (9 by default), the values regulating number of conformations (number of conformations for a fragment, number of candidates, and number of conformations to keep after each “join”) are increased by a factor that is a function of the number of the rotatable bonds.
Conformations are ranked using a sum of Lennard-Jones 6-12 non-bonded potentials (for rapid processing the L-J parameters are obtained from a look up table of DREIDING force-field [6] values), ad-hoc tabulated dihedral penalties associated with the inter-fragment bonds, and contributions that facilitate equatorial (versus axial) ring attachment configurations by enhancing specific 1-4 interactions. The process of joining progressively larger fragments together and sampling multiple orientations for the restored bond continues until the entire molecule is reconstructed. Duplicate conformer elimination is only performed for molecules smaller than a certain threshold (having less than 32 heavy or polar hydrogen atoms by default) because this process is slow and rarely impactful for the large molecules.
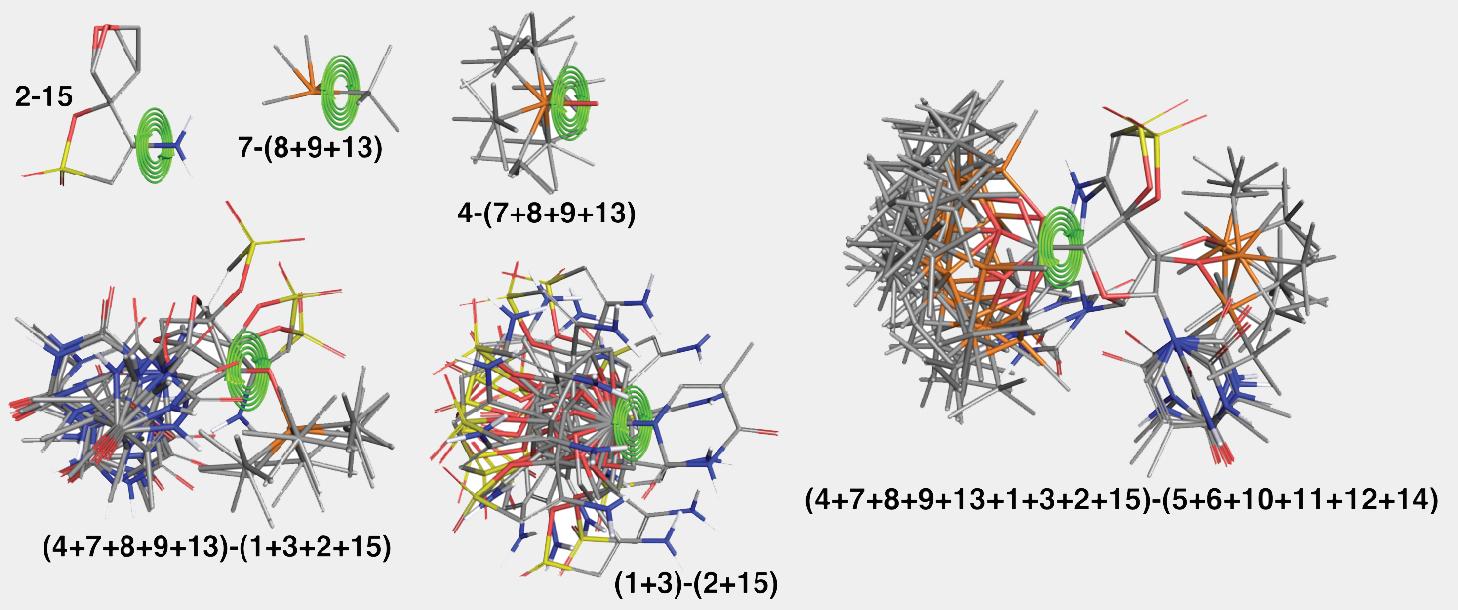
Key steps of the algorithm are illustrated in Figure 2 and Figure 3 for the ligand from PDB structure with code 3QO9. The structure is split into fifteen fragments as shown in Figure 2. Only the fragment labeled as “2” has more than one low-energy conformation. The algorithm picks the order in which fragments are assembled to minimize the number of the atom pairs that would need to be checked for overlaps. For this structure the order was as follows: 1+3, 2+15, 5+6, 9+13, 12+14, 8+(9+13), 11+(12+14), (1+3)+(2+15), 7+(8+9+13), 10+(11+12+14), 4+(7+8+9+13), (5+6)+(10+11+12+14), (4+7+8+9+13)+(1+3+2+15), (4+7+8+9+13+1+3+2+15)+(5+6+10+11+12+14). Many assembly steps involving simple fragments, e.g., 1+3, 5+6, 12+14, etc., resulting in a merged fragment that has only a single distinct conformation. Illustrative stages of the assembly algorithm are shown in the Figure 3.
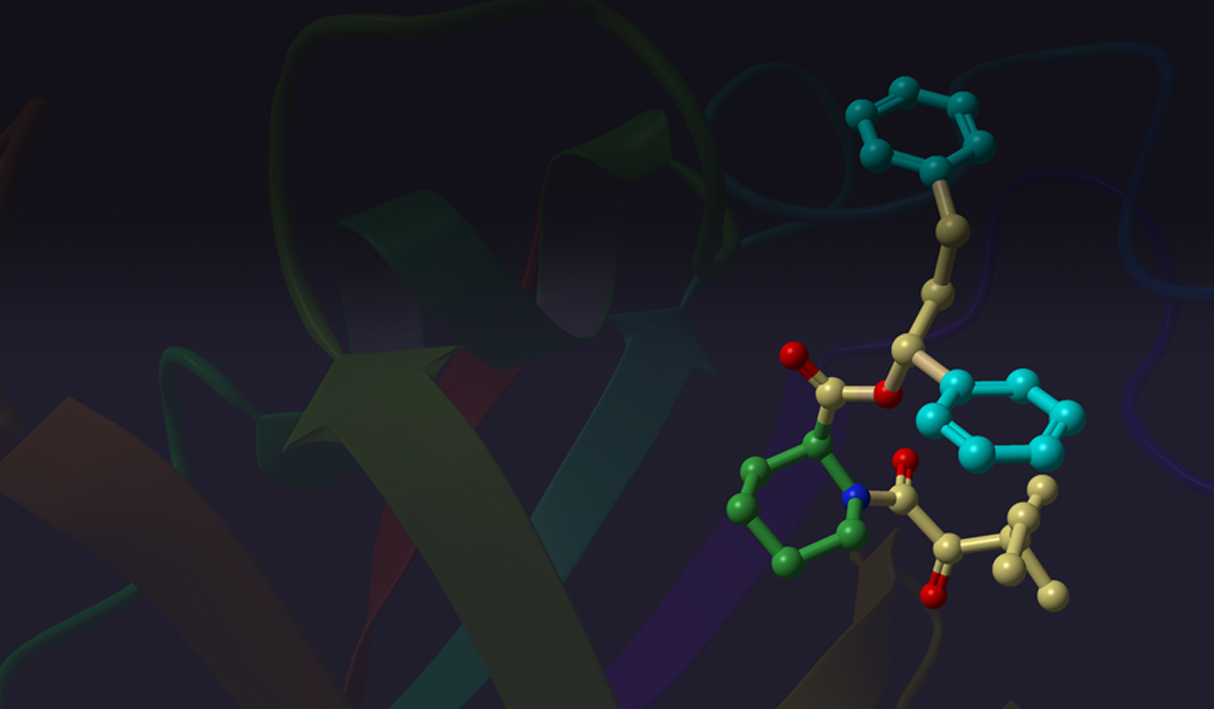
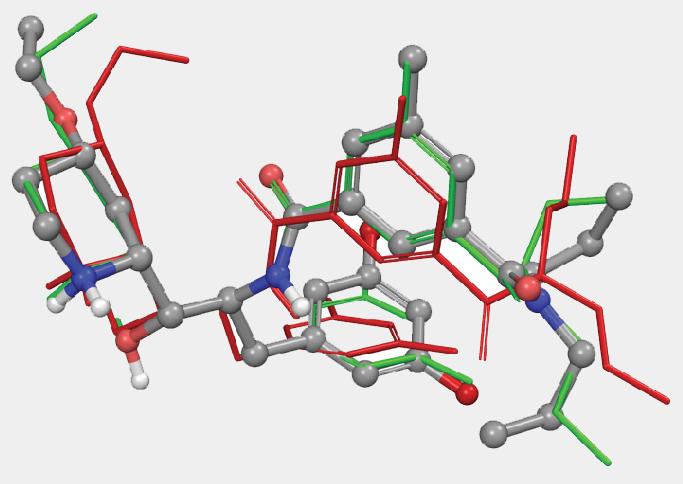
Several ConfGen parameters can be used to control tradeoffs in calculation speed and recovery of bioactive conformations. The two most important are: whether the geometry of the conformers is optimized using an OPLS force field and the number of conformers the program is expected to generate. The default dielectric constant of 10 used during minimization was found to generate bioactive conformers better than using 1. A real-world example is shown in Figure 4. Apart from resolving bad atomic contacts, geometry optimization typically leads to mildly improved bioactive conformation recovery (see Table 1). Optimization, however, is about 20 times slower than running without minimization. Optimization also tends to reduce the number of conformers; something that can lead to reduced processing time in subsequent calculations that use these conformations.
Another parameter controlling the speed/quality tradeoff is the number of conformers to be generated per input ligand (-n command line option): larger values of this parameter may often result in better conformer diversity. This is illustrated in Figure 4 for the ligand from 2QP8 PDB entry (1.5Å resolution): using default simulation settings, the conformer closest to the bioactive conformation has an RMSD of 1.3Å (red). The RMSD drops to 0.6Å (green) if the maximum number of conformers is increased from 64 (default) to 256, and geometry optimization using OPLS3 force-field is enabled.
Conclusions
Given its exceptional speed and ability to reproduce low RMSD bioactive conformations, ConfGen is the ideal solution for generating conformations for ligand-based virtual screening. It is tightly integrated within Maestro and Schrödinger’s ligand-based drug discovery tools, including Phase, which provides the ability to easily create databases for fast ligand-based virtual screening. As with all Schrödinger products, ConfGen is also constantly being refined and improved, so while the updates described in this paper represent a significant step forward compared to previous iterations, they will also act as the baseline for future enhancements.
References
-
Friedrich, N.O.; et al., “Benchmarking Commercial Conformer Ensemble Generators,”
J. Chem. Inf. Model,” 2017, 57(11), 2719-28.
-
Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G., “ZINC: A Free Tool to Discover Chemistry for Biology,”
J. Chem. Inf. Model., 2012, 52(7), 1757-68.
-
Pyykkö, P.; Atsumi, M., “Molecular Single-Bond Covalent Radii for Elements 1-118,”
Chemistry, 2009, 15(1), 186-97.
-
Pyykkö, P.; Atsumi M., “Molecular Double-Bond Covalent Radii for Elements Li-E112,”
Chemistry, 2009, 15(46), 12770-9.
-
Alvarez, S., “A Cartography of the van der Waals Territories,”
Dalton Transactions, 2013, 42(24), 8617-36.
-
Mayo, S.L.; Olafson, B.D.; Goddard, W.A., “DREIDING: A Generic Force Field for Molecular Simulations,”
J. Phys. Chem., 1990, 94, 8897-8909.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.