Improving the accuracy of protein thermostability predictions
We sat down with Dr. Jianxin Duan to discuss his paper, “Improving the Accuracy of Protein Thermostability Predictions for Single Point Mutations” which recently appeared in the Biophysical Journal.
Tell us what was the main motivation behind this study?
The function of a protein is tightly coupled with its structure and dynamic behavior. Therefore, understanding the thermostability of proteins can provide fundamental insights into how they work. For example, single point missense mutations may directly affect protein function, leading to diseases. Many of these mutations have been found to be linked to protein thermostability. Also, from a practical perspective, protein stability engineering has wide applications in various industries, for example, vaccines and antibodies need to be engineered to have long shelf life and prevent aggregation; industrial enzymes for food, detergents, paper, or fuel need to be designed to be stable and functional in the desired environments.
As you know, we’ve had tremendous success in using our FEP+ technology in advancing small molecule drug discovery—we’ve published many papers, and all of our drug discovery projects have achieved milestones at an accelerated pace when compared to traditional approaches. But what’s less well established is how well FEP+ could advance projects involving biologics, so we set out to design a study to specifically examine the performance of FEP+ in predicting protein thermostability. We aimed to assess both quantitatively and qualitatively the technology as a viable option to predict changes in protein thermostability upon mutations such that it may guide protein engineering projects.
Had there been previous studies using FEP on proteins? And if so, how does the current study differ?
The idea of applying free energy perturbation theory (FEP) to predict the effect of single point mutations on protein stability is not new; however, production-level calculations to predict protein stability in industrial settings have not been possible due to a couple of severe limitations, including the inability to model mutations to and from prolines, where the bonded topology of the backbone is modified, and the complexity in modeling charge-changing mutations. In this study, we extended the FEP+ protocol to accurately model both of these types of mutations. Furthermore, because FEP calculations could be computationally intensive, many earlier studies were limited to very short simulations, and with limited solvation around the mutation, ultimately resulting in less accurate predictions. Thanks to algorithmic improvements and hardware advances in GPUs, Schrödinger’s FEP+ has demonstrated repeatedly that it can meaningfully impact projects within realistic timelines.
Because we set out to specifically assess FEP+’s performance in dealing with the challenging proline and charge-changing mutations, we paid particular care to selecting the test systems so that we could quantitatively evaluate our results – we curated a data set with high-resolution crystal structures for both wild-type and mutants, and with accurately measured stability data. You may find details of our test systems in the paper. And I’m happy to report that we achieved comparable accuracy in predicted stability changes as observed previously in countless small molecules studies, demonstrating that FEP+ is indeed an appropriate computational strategy for studying biologics.
Are there other methods for predicting protein thermostability? And if so, how do the present FEP+ results compare?
Yes, because of the importance of being able to accurately predict protein stability, many different methods have been proposed over the years. We compared our FEP+ results with several of the more popular methods.
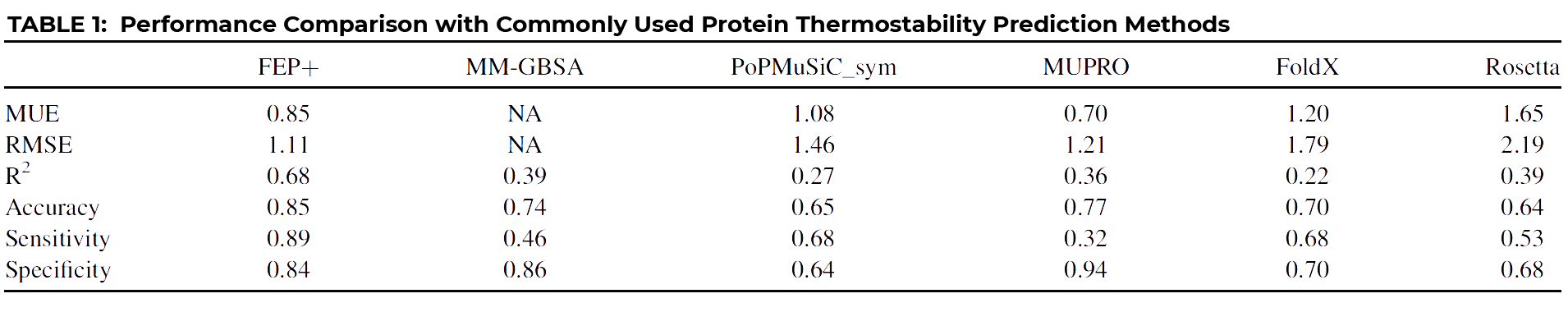
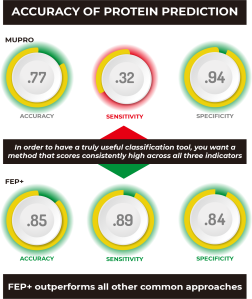
As you can see at a glance, FEP+ outperforms all other common approaches quantitatively, with the possible exception of MUPRO, which is a machine learning model using only sequence information and all mutations as part of its training set. However, it’s been suggested that MUPRO may suffer from training set bias, and indeed this is born out when you examine the qualitative measures of Accuracy, Sensitivity, and Specificity, also given in the table.
Let me explain what these three measures mean by using an example:
- if experimentally it’s found that 90 out of 100 mutations are destabilizing, and one blindly guessed that all mutations are destabilizing,
- then one’s guess would score 90% for Accuracy,
- but would score 0% for Sensitivity, which measures the ability to correctly identify stabilizing mutations,
- and 100% for Specificity, which measures the ability to correctly identify destabilizing mutations.
In order to have a truly useful classification tool, you’d want a method that scores consistently high across all three indicators, like FEP+, which clearly outperforms all other methods, including MUPRO, which while scoring well for Specificity, owing to its training set, it nevertheless is biased by that training set to be a poor predictor of stabilizing mutations, as indicated by its very low Sensitivity score of 0.32.
But isn’t FEP+ computationally expensive? How well can it be deployed at scale to meaningfully accelerate a discovery program?
It is true that FEP+ studies require a computational investment, but thanks to software algorithmic improvements and the ever-increasing processing power of GPU hardware, full analyses using FEP+ is well within reach for real-life projects. Furthermore, as described in our paper, we devised a screening cascade taking advantage of the much more computationally efficient residue scanning, available within our BioLuminate application, which tends to err on the side of false positives, as a filter for further FEP+ analyses. This way, one could rapidly screen all relevant mutations, and only pass onto FEP+ those that are found to be stabilizing, and let FEP+ identify those mutations that are most likely to be true positives.
That all sounds very promising, are there future plans for enhancing or expanding the technology?
Yes, of course, we’ve learned a great deal from this study, such as how to best model the protein’s unfolded state to achieve higher accuracy in the predicted change in stability. We also scrutinized our results and identified potential cases that may require longer simulations to fully capture large structural changes. We will continue to refine the protocol to improve the performance both in terms of prediction throughput and accuracy.
Author

Jianxin Duan
Director, Applications Science, Schrödinger
Jianxin Duan, Ph.D., is a Director with Schrödinger’s Applications Science group. He earned his Ph.D. in Biophysics from Karolinska Institute in Stockholm. Dr. Duan has been with Schrödinger for 14 years. He was involved in the development of ligand based methods, including cheminformatics, pharmacophore modelling and machine learning. His current personal research focuses on protein and antibody design.