JUN 12, 2025
Modeling Molecular Scale Dynamics of Kinetically Gated Carbon Dioxide Capture Using Photoswitch Functionality in Metal Organic Frameworks
Abstract:
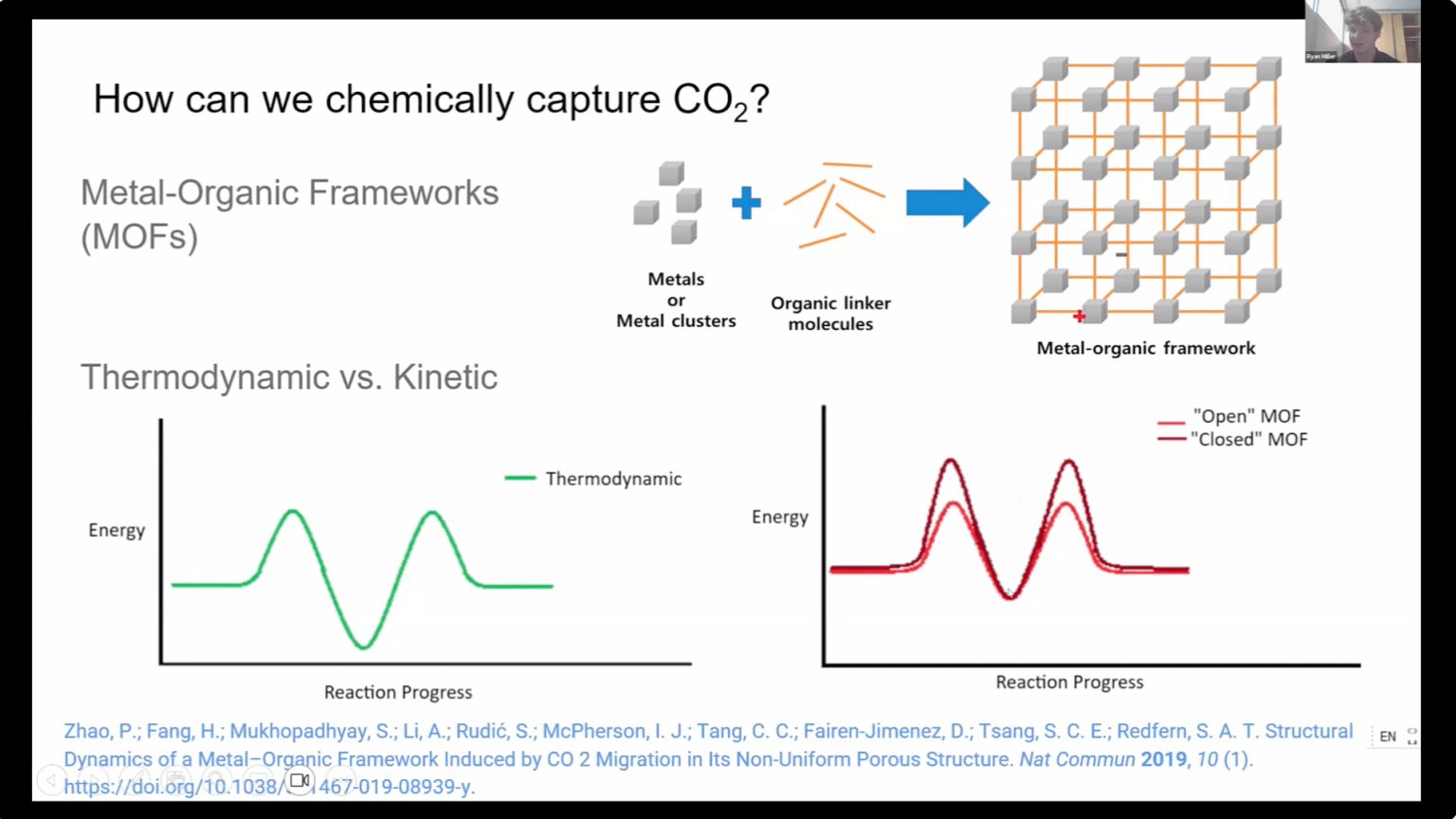
Carbon emissions are increasingly causing concern due to their effects on climate. Metal–organic frameworks (MOFs) are promising materials that have been shown to be effective for low-energy carbon capture. This study focuses on MOFs with the addition of azobenzene moieties to kinetically control carbon dioxide (CO₂) transit. The spatial conformation of azobenzene can be switched between the cis and trans conformation upon interaction with light. These molecular changes influence pore accessibility, enabling tunable diffusion rates of CO₂ without the energy costs associated with thermodynamic modulated MOF capture.
Using computational molecular dynamics simulations, single molecule CO2 transit was modeled through UiO-68-MOFs with azobenzene photoswitches in three configurations: trans (open), cis (closed), and no switch. The initial geometry of the MOF was optimized with the ORCA quantum chemistry program, and MD parameters were derived using tinker AMOEBA software. Simulations were run using OpenMM software, with periodic boundary conditions. The CO2 transits were modeled as a multi-step kinetic process that was simulated using a Markov chain stochastic approach.
The simulations revealed significant differences in CO₂ transit. Trans switch conformations yielded faster passage compared to cis, with average transit rates 33% higher. Statistical analysis confirmed these differences were significant, particularly across key transitions into and out of the MOF pore. These findings align with experimental work and support the hypothesis that photoswitch modulation has the potential to kinetically gate CO₂ flow.
Induced dipole interactions with copper vertices were also noted to differ based on switch conformation, but need further work to determine if they contribute to transit rate differences. Future research will investigate differing metal ions and structural changes to the azobenzene moieties. Activation energies will be calculated through temperature-dependent simulations to maximize the difference between open and closed conformations. These insights lay foundational groundwork to design MOFs for low-energy carbon sequestration technologies.
Presenter

Ryan Miller
Pacific University
Ryan Miller is from Hillsboro, Oregon, and recently received his Bachelors in Chemistry from Pacific University. He has conducted computational chemistry research with Dr. Kevin Johnson researching materials for carbon capture