Machine learning force fields for improved materials modeling
Machine learning force fields (MLFFs), also known as machine learning interatomic potentials, represent an intermediate between classical force fields and density functional theory (DFT), maintaining the linear scaling of the former while approaching the accuracy of the latter. Beyond the balance of accuracy and efficiency/cost, MLFFs are enabling new scientific insights by making large-scale and longtime scale simulations feasible for reactive systems. This opens the door to modeling complex materials systems that were previously computationally prohibitive with traditional quantum methods.
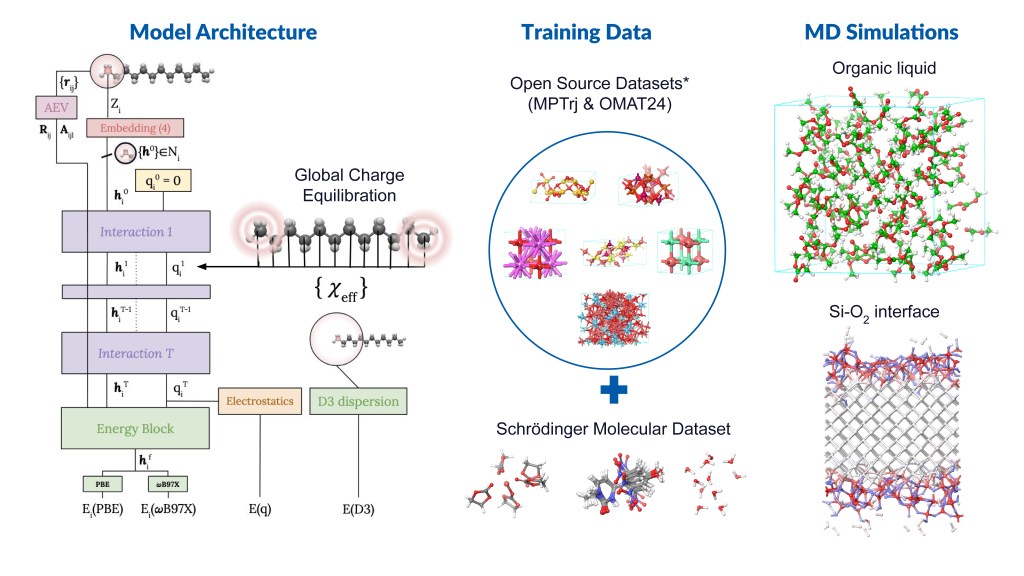
Message Passing Network with Iterative Charge Equilibration (MPNICE) is an MLFF architecture developed by Schrödinger for which multiple pretrained models spanning 89 elements are available, and which explicitly incorporates equilibrated atomic charges and long range electrostatics.1 This technological advancement has removed the drawback of previous MLFFs that were limited by the number of unique atomic elements they could model. Furthermore, inclusion of atomic charges and electrostatics through charge equilibration has enabled representation of multiple charge states, ionic systems, and electronic response properties, while simultaneously improving accuracy. In addition to MPNICE, the Schrӧdinger suite also allows users to utilize the Universal Models for Atoms (UMA),2 developed at Meta. This suite of models offers very high accuracy, includes a model that yields good performance for reaction barrier heights for finite systems, and covers the majority of the periodic table.
By integrating state-of-the-art MLFF methods with high-performance OPLS4 or OPLS5 force fields, as well as advanced DFT and molecular dynamics (MD) engines, Schrödinger offers a uniquely powerful platform for materials simulation — positioning us as the leading partner in advanced MLFF technologies. In this application note, we present case studies from materials-intensive industries, including batteries and catalysis.
Benefits of MLFF
Near DFT-level accuracy with orders of magnitude reduction in computational time
Option for GPU accelerated molecular dynamics with Desmond
Large chemical space spanning 89 elements
Specialized force fields for organic, inorganic, and hybrid materials
Diverse applications of MLFF
Batteries:
- Calculate bulk and transport properties, such as diffusion, viscosity, and conductivity of liquid electrolytes
- Simulate Li-ion diffusion in solid-state electrolytes and cathode coating materials
- Model electrolyte reactivity and SEI formation
OLED materials:
- Simulate molecular packing and thin-film morphology
- Investigate doping, host-guest, and interlayer interactions
- Link device properties to the static and dynamic disorder of molecular systems
- Facilitate thermomechanical property prediction
- Model charge and exciton transport
Crystal structure prediction:
- Rank order organic crystal structures
Adsorption on surfaces:
- Study reactivity of multiple adsorbates in extended models of complex surfaces
Reactivity in molecules and solid-state:
- Investigate reaction pathways and transition states
- Expedite catalyst design
- Optimize chemical reactions
Polymers:
- Evaluate polymer dynamical properties
- Investigate solid polymer electrolytes
Read the full white paper
References
-
Efficient long-range machine learning force fields for liquid and materials properties.
Weber JL, et al. arXiv:2505.06462. 1 Aug 2025.
-
UMA: A Family of Universal Models for Atoms.
Wood BM, et al. arXiv:2506.23971. 30 June 2025.
-
Synthesis of lithium aluminate for application in radiation dosimetry.
Ha, NTT, et al. Materials Letters. 2020, 267, 127506.
-
Ultrathin lithium-ion conducting coatings for increased interfacial stability in high voltage lithium-ion batteries.
Park, JS, et al. Chemistry of Materials. 2014, 26, 3128–3134.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.