A design strategy of exciton blocking materials using simulations and the analysis of device properties
Steric Rooted Multi-Resonant Thermally Activated Delayed Fluorescent Emitters for Pure Blue Organic Light Emitting Diodes with Ultralow Efficiency Roll-Off
Thiazoline Carbene−Au(I)−Amide complexes exhibiting dimer electroluminescence
In silico enabled discovery of KAI-11101, a preclinical DLK inhibitor for the treatment of neurodegenerative disease and neuronal injury
QuickShape screening in the age of ultra-large libraries
QuickShape screening in the age of ultra-large libraries
Tim Knehans, Steve Dixon, Nick Boyles, Jianxin Duan, Chris von Bargen, Volodymyr Babin, Steve Jerome, Matt Repasky
Readily available chemical libraries continue to increase rapidly in size while the need for novel chemical matter for drug targets remains at an all time high. Although 3-dimensional Shape-based methodologies provide an effective approach to enrich actives by finding hits that are topologically different but similar in shape, their hardware requirements can become prohibitive for screening ultra-large chemical libraries. Here we compare the performance of a 1-dimensional (1D) pharmacophore-based fingerprint with GPU-based Shape Screening both in terms of enrichment of known actives as well as computational speed.
The 1D method not only retains enrichment at levels very similar to Shape Screening with high overlap of recovered actives but also provides distinct hits. Additionally, it allows screening of billions of molecules on a conventional laptop within a few days and requires 100 times less storage. By combining both 1D and 3D approaches, we developed an automated staged workflow called QuickShape enabling efficient 3D-Shape screenings for library sizes in the tens of billions of molecules. This workflow is an excellent ligand-based screening solution in the new era of ultra-large libraries.
1D-fingerprint technology enables pre-screening of Enamine REAL in a few hours within QuickShape workflow
QuickShape is a cascaded screening workflow using 1D pharmacophoric fingerprint screening followed by Shape Screening. Pharmacophores are perceived for each molecule with appropriate protonation and tautomer states and subsequently projected into a fingerprint vector. The distance between the pharmacophoric points is the number of bonds between the pharmacophores.
This process takes approximately 1 CPU hour per 1 million molecules. The output database requires 100 times less storage compared to equivalent Shape GPU databases. A 6.5 billion compound database takes up only 400 GB.
The similarity is defined by comparing the overlap of equivalent pharmacophores between the molecules A and B:
Sim(A, B) = Overlap(A, B)/max[Overlap(A, A), Overlap(B, B)]
The similarity comparison is extremely fast and parallelizable. It is possible to screen 6.5 billion compounds on a computer with 10 cores in 14 hours.
The hits from 1D screening are triaged using Shape Screening and the final hit list can be analyzed based on diversity, specific pharmacophore criteria and property filters using the Hit Analyzer panel in Maestro. The complete workflow can be run on a laptop in a few days.
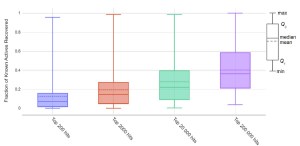
1D Screening provides similar enrichment compared to Shape screens
First we validated the enrichment of 1D similarity against that of Shape using the actives from the DUD-E dataset seeded in 1.4 billion molecules from Enamine REAL. The five most diverse actives were used as probes. Both methods provide very similar enrichment of known actives. Among top 200,000 hits, actives were identified for all targets, indicating 1D screening alone can be useful. It is important to note that real enrichment may be much higher because there are likely additional unknown actives in the Enamine REAL database.
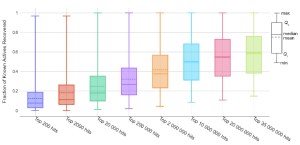
1D Screening & Shape have good correlation and high overlap
Secondly, we ensure that 1D screening is able to retrieve the same molecules as Shape, which is represented by the percent overlap among the top hits. Among the top 20 million hits from 1D screening we find a minimum 50% and on average 90% of the top 200,000 hits from Shape screening. Already at 200,000 returned hits, the average overlap is >60% indicating that it may be sufficient to run 100 times less Shape calculations.
The 1D similarity score is highly correlated with Shape similarity with an R2 of 0.65. This demonstrates that 1D screening is able to reduce the number of Shape comparisons by 84% while maintaining similar performance.
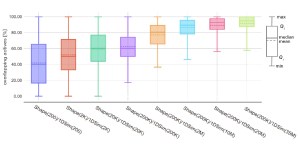
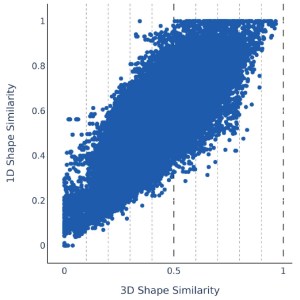
QuickShape and its constituent technologies should be used in a combined fashion
Since the enrichment between Shape and 1D screening (1DSIM) are similar but neither correlation nor overlap are 100%, we visualized the trends in unique contribution to the recovered actives by 1D Screening via calculating:
#actives1DSIM – 2 x #actives1DSIM ∩ Shape
In this representation, all values above 0 show targets where at least twice as many actives uniquely returned by 1D screening compared to overlapping hits. As with previous figures the more molecules are returned by each methodology the bigger the overlap gets and the distributions are shifted below 0 which makes these molecules accessible to the QuickShape workflow. However, in all bins, significant amounts of molecules are above and close to 0. This indicates the ability of 1D screening to contribute valuable hit matter on its own hence it is generally recommended to investigate the top-ranked molecules by either methodology in order to increase the likelihood of novel hit identification.

Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Modeling Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
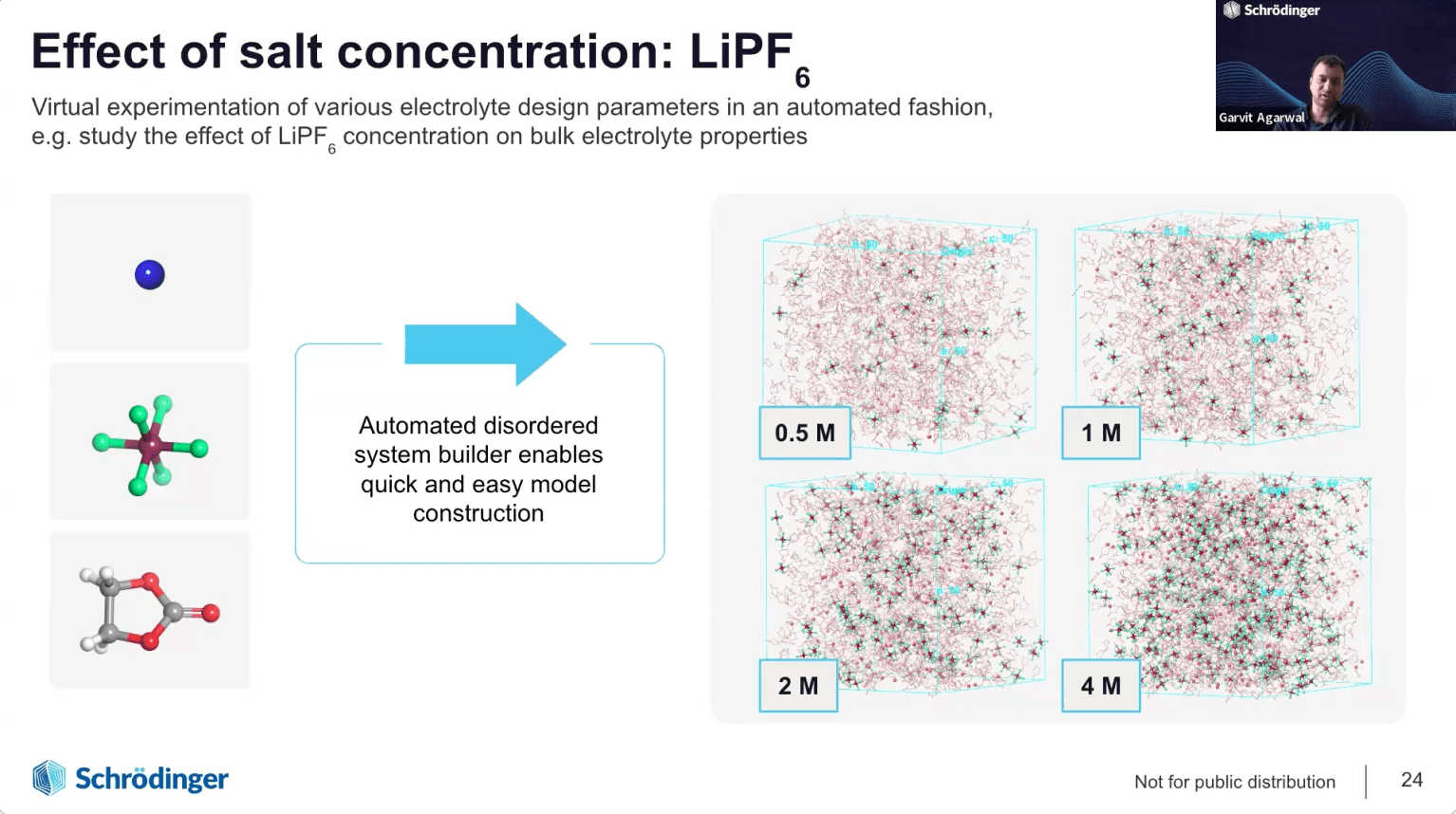
Electrodes, electrolytes & interfaces: Harnessing molecular simulation and machine learning for rapid advancements in battery materials development
JUN 26, 2024
Electrodes, electrolytes & interfaces: Harnessing molecular simulation and machine learning for rapid advancements in battery materials development
Schrödinger is excited to be presenting in a webinar hosted by the Battery Technology Platform, taking place on June 26th. Join us for a presentation by Garvit Agarwal, Ph.D., Scientific Lead at Schrödinger, titled “Electrodes, electrolytes & interfaces: Harnessing molecular simulation and machine learning for rapid advancements in battery materials development.”
Abstract:
Li-ion battery (LIB) technology has revolutionized industries like transportation and consumer electronics. However, new battery chemistries are needed to address rapidly growing demand and to improve the power density, safety, reliability, and lifetime of LIBs.
In this webinar we will explore the key materials challenges for improving battery performance and demonstrate how atomistic simulation and machine learning (ML) enable swift evaluation and screening of vast design spaces, accelerating the introduction of innovative technology to market.
We will introduce the latest technological innovations in Schrödinger’s digital chemistry platform for battery materials design. In particular, the webinar will focus on examples to demonstrate the application of automated solutions for accurate prediction of thermodynamic stability and voltage profile of cathode, ion diffusion pathways and kinetics in electrode materials, transport properties of liquid electrolytes and modeling the nucleation and growth of solid electrolyte interphase (SEI) layers using Schrödinger’s SEI simulator module. We will also introduce an automated generalized framework for the development of customized machine learning force fields for complex materials such as liquid electrolytes, inorganic cathode coatings and solid polymer electrolytes, paving the way for efficient design of novel materials for next generation batteries.
Key Learning Objectives:
- Gain insight into how a digital chemistry approach reduces development cycle time for new battery materials
- Learn how Schrödinger’s automated high throughput simulation workflows and ML models enable rapid screening of battery materials candidates
- Learn how to leverage automated solutions for accurate prediction of key battery properties: thermodynamic stability and voltage profile of electrodes, ion diffusion pathways and kinetics in electrodes, transport properties of liquid electrolytes, and the nucleation and growth of solid electrolyte interphase (SEI) layers
- Hear applications of advanced machine learning force fields for accurate modeling of electrolyte materials, cathode coatings and interfaces

Garvit Agarwal, Ph.D.
Scientific Lead, Energy Storage Materials Science Group, Schrödinger
Garvit Agarwal is Senior Scientist and Scientific Lead for Energy Storage at Schrödinger, working to extend and apply molecular modeling tools for the accelerated discovery of next-generation clean energy technologies. Garvit obtained his Ph.D. in Materials Science and Engineering from the University of Connecticut. He worked as a post-doctoral researcher in the Materials Science Division at Argonne National Laboratory prior to joining the Materials Science team at Schrödinger.