Macromolecular refinement of X-ray and cryo-electron microscopy structures with Phenix / OPLS3e for improved structure and ligand quality
Toward in vivo-relevant hERG safety assessment and mitigation strategies based on relationships between non-equilibrium blocker binding, three-dimensional channel-blocker interactions, dynamic occupancy, dynamic exposure, and cellular arrhythmia
Performance and Its Limits in Rigid Body Protein-Protein Docking
Novel, Self-Assembling Dimeric Inhibitors of Human ‘ Tryptase
Digitalisierung: molekulares Design plattformisieren
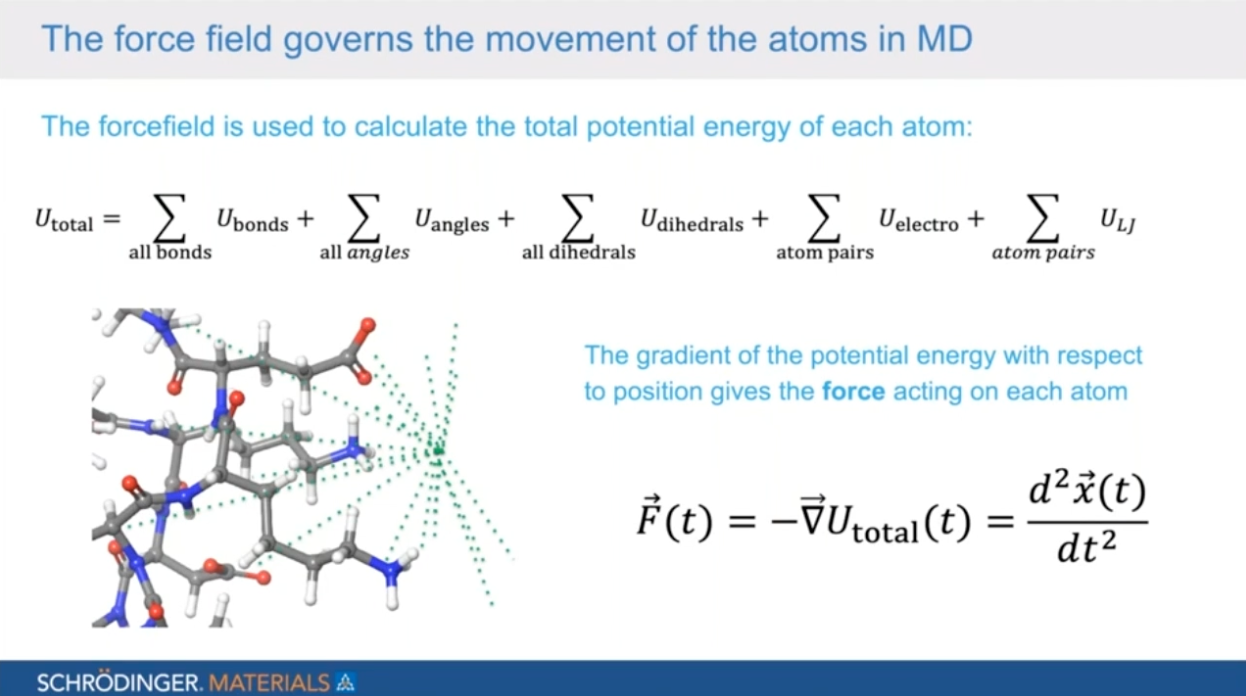
Surfactant chemistry development for consumer packaged goods enhanced by atomic scale simulation

AUG 10, 2020
Surfactant chemistry development for consumer packaged goods enhanced by atomic scale simulation
Speaker:
Jeffrey Sanders, Principal Scientist
Abstract:
Surfactants play a key role in formulations from emulsifiers in candy bars to home detergents. The design of new chemistries and the development of novel formulations can be aided by predicting and understanding the link between chemical structure and performance. Atomic scale simulation provides an increasingly powerful avenue to explore the impact of chemistry and formulation changes in the performance of surfactant-based chemistries. In this webinar, we will present case studies on how physic-based chemical simulation in the Schrödinger Materials Science Suite can be used to help predict and rationalize performance determining properties of surfactants for the CPG market applications.
Learning to Taste: Application of Deep Learning to Predict the Sweetness of Small Organic Molecules


AUG 6, 2020
Learning to taste: Application of deep learning to predict the sweetness of small organic molecules
Speaker:
Atif Afzal, Senior Scientist
Abstract:
With the recent developments in machine learning techniques, we use the data available on the taste of existing chemistries to develop efficient data-driven models for taste prediction. We demonstrate that these advanced machine learning models are highly efficient in classifying the molecules as bitter/sweet. Using the data and the model developed in this work, we can not only predict the sweetness of new molecules but also identify underlying relationships that distinguish the molecules as bitter/sweet.
Perspectives in Computational Materials Design: Progress and Prospects
AUG 6, 2020
Perspectives in computational materials design: Progress and prospects
Speaker:
William A. Goddard, III
Abstract:
New strategies for multiparadigm simulations of nanoscale materials with applications to electrocatalysis, Li batteries, micelle formation, ductile boron carbide, etc.
Controllable Singlet-Triplet Energy Splitting of Graphene Quantum Dots through Oxidation: From Phosphorescence to TADF
Computer-aided Formulation Development for Small-molecule Drugs


JUL 16, 2020
Computer-aided Formulation Development for Small-molecule Drugs
Speaker
Dr. Shaun Kwak
Senior Principal Scientist and Applications Lead
Abstract
With the accelerating pace of drug discovery, fast and efficient ways to both pre-formulate and formulate new drugs have become critical elements of the pharmaceutical development. The latest advancements in atomistic-scale modeling and simulation technology have enabled in silico screening capability through a large number of candidate materials and formulations, based on fully physics-based models. In this webinar, we will discuss the key areas of formulations science powered by the latest computation chemistry technology, including analysis of chemical processes, assessment of intermolecular interactions in particle/drug systems, and automated in silico property characterization.
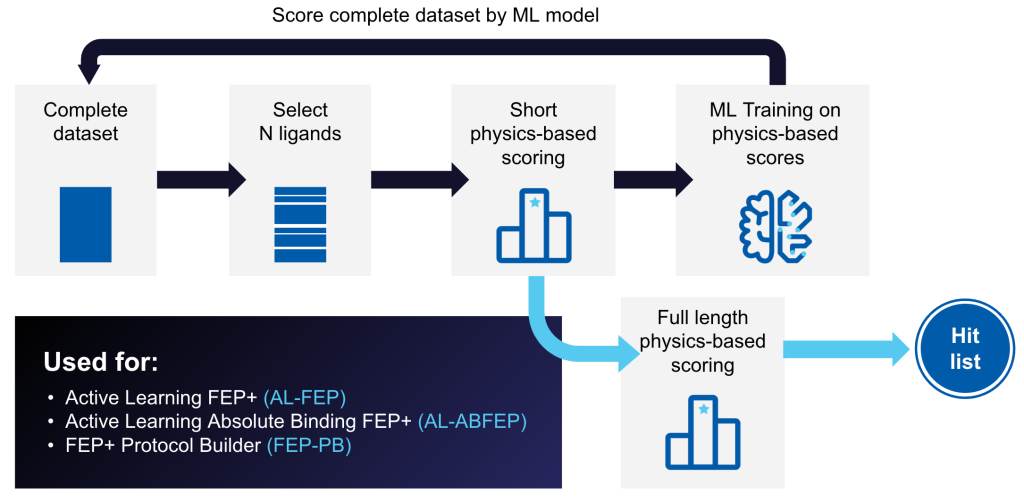
Active Learning Glide – Screen Billions of Compounds Efficiently and Cost Effectively

JUL 13, 2020
Active Learning Glide – Screen Billions of Compounds Efficiently and Cost Effectively
Speaker
Dr. Matt Repasky
Vice President, Life Sciences Products
Abstract
Docking has proven its value identifying novel hits from purchasable compound libraries. With the advent and rapid growth of made-on-demand libraries, purchasable compound space has grown from ~10 million compounds to well over a billion compounds. We illustrate how using an Active Learning approach combined with Glide enables cost effective and accurate screening of billion compound libraries. Application to A2A and SRC targets finds at least 70% of the best 1000 ligands from brute force docking of a 100 million compound library are found in the top-ranked 1000 ligands by ActiveLearning Glide followed by Glide redocking of 1 million compounds.