JUL 8, 2020
Enumeration as a computational strategy for automating the design of CVD and ALD precursors
Speaker:
Simon D. Elliott, Director, Atomic Level Process Simulation
Abstract:
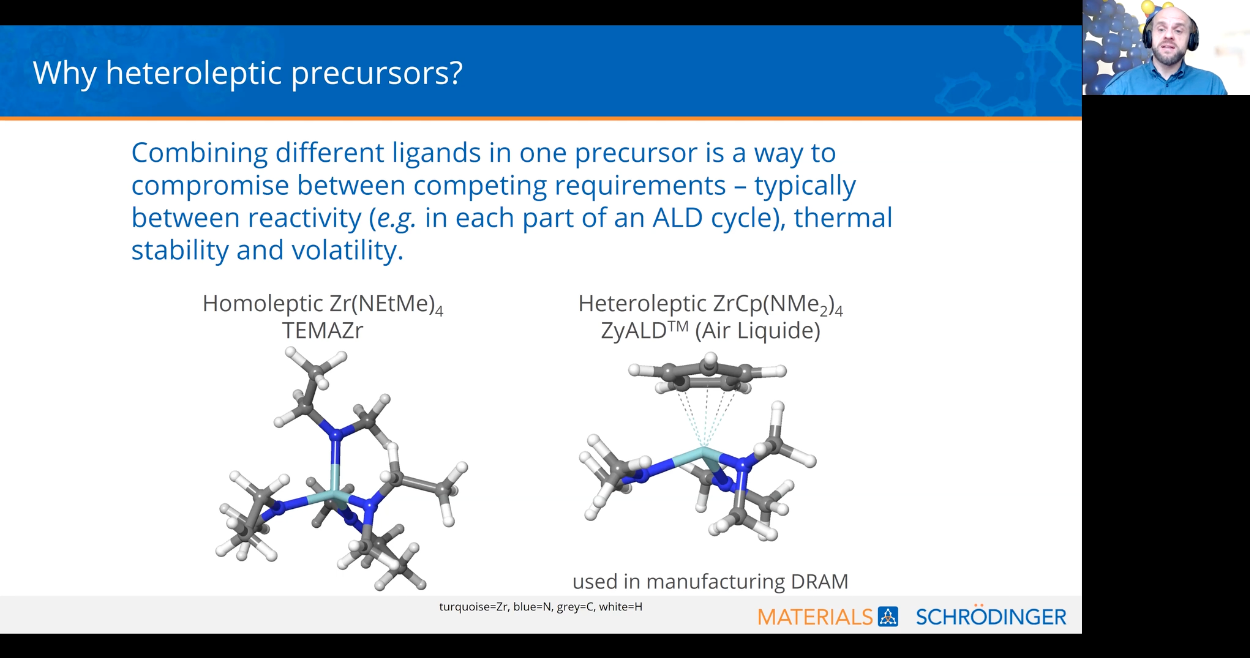
The success of chemical-based deposition and etch depends primarily on the choice of gas-phase chemicals. For atomic layer deposition (ALD) and chemical vapor deposition (CVD) more generally, the precursor chemicals are often metalorganic complexes with ligands surrounding a metal center. Ligand choice in ALD/CVD precursors is crucial for throughput, stoichiometry, impurities, and process temperature. Heteroleptic precursors (containing more than one type of ligand) are one way to compromise between conflicting chemical requirements. However, to date we have barely ‘scratched the surface’ of the vast chemical space of possible heteroleptic precursors. Since an exhaustive experimental analysis is not possible, we look to computational screening to narrow down the search to the most promising options. Here we present a computational approach for screening metal precursors with respect to thermal stability. The computational strategy is illustrated on the example of Zr precursors for zirconium nitride, used as a hard coating to protect industrial parts in corrosive environments.