PIPER
A state-of-the-art protein-protein docking program

A state-of-the-art protein-protein docking program
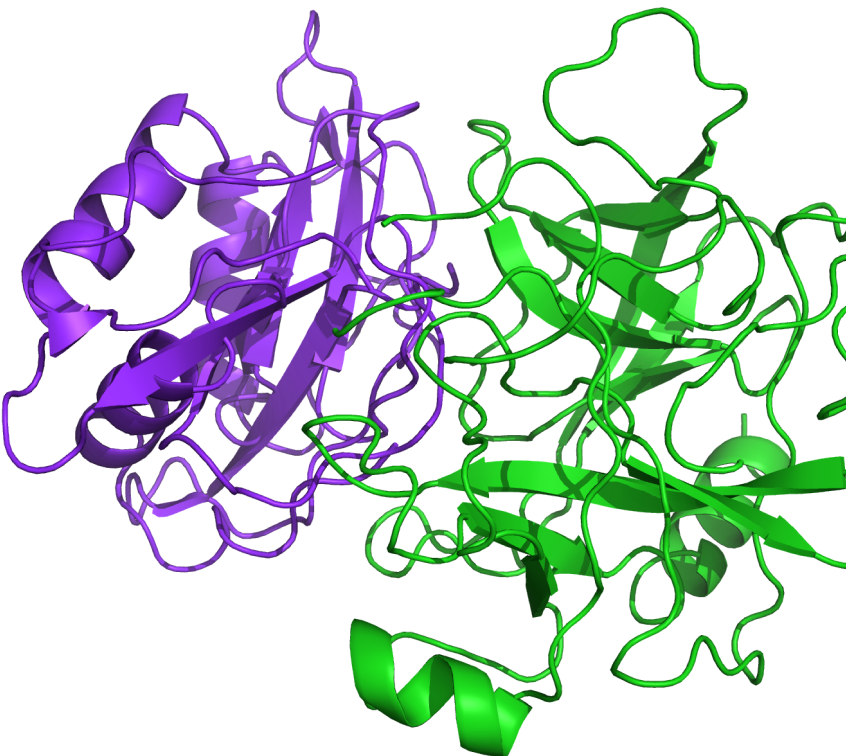
PIPER is a well-validated protein-protein docking program based on a multi-staged approach and advanced numerical methods that generates reliable structures of protein-protein complexes. Based on docking code from the Vajda lab at Boston University, PIPER has a proven track record as an outstanding predictor of protein-protein complexes as judged by previous CAPRI (Critical Assessment of Prediction of Interactions) blind experiments.
Get answers to common questions and learn best practices for using Schrödinger’s software.

Learn more about the related computational technologies available to progress your research projects.
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.