Crystal Structure Prediction (CSP)
De-risk your solid form selection process by identifying the most stable polymorph at room temperature

De-risk your solid form selection process by identifying the most stable polymorph at room temperature
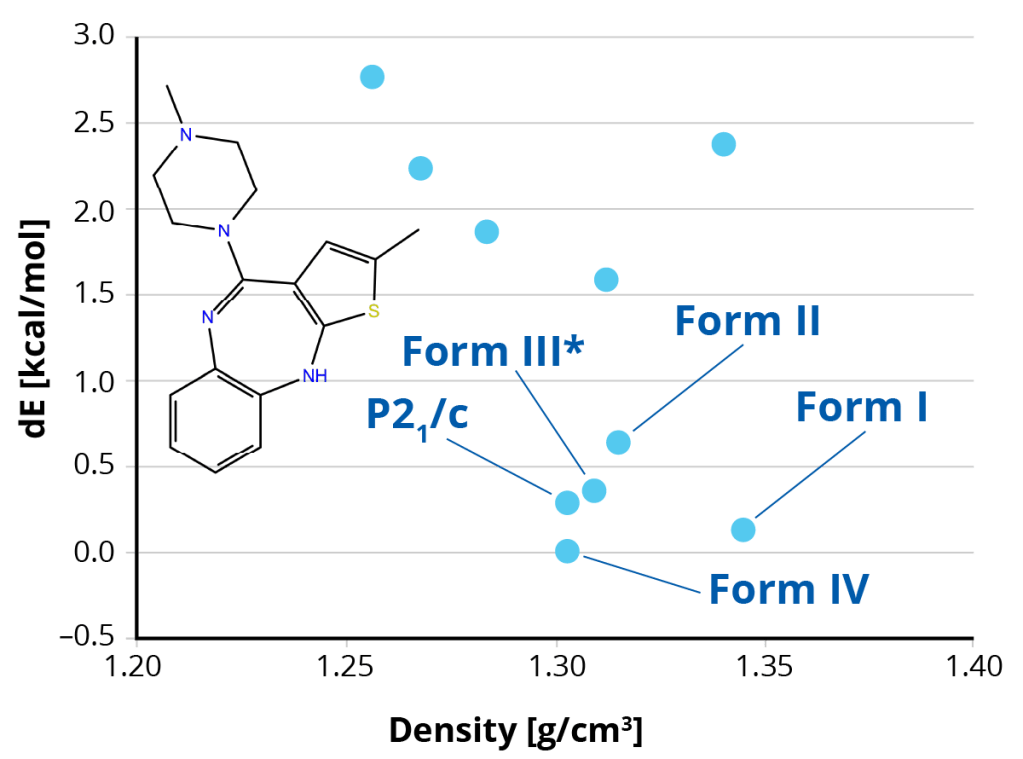
Overcome the risks associated with disappearing polymorphs in late stage drug development. Schrödinger’s proprietary crystal structure prediction (CSP) platform identifies the stable crystal polymorphs at 0K and RT for a given active pharmaceutical ingredient (API). The CSP platform can now be used to predict crystal polymorphs in the Z’=2 search space, salts and monohydrate polymorph systems.

Optionally predict key properties of an API to support selection of a stable solid form.

Work with our team of computational experts to de-risk your solid form selection process. Starting from a 2D structure of the API, Schrödinger’s team will deliver to you the thermodynamic stability ranking of crystal polymorphs.

Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Zhou D, et al. Nature Communications, 2025, 16, 2210
Hong RS, et al. J. Med. Chem. 2023, 66, 23, 15883-15893
Hong RS, et al. J. Chem. Inf. Model. 2021, 61, 3, 1412-1426
Learn more about the related computational technologies available to progress your research projects.
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
Integrated graphical user interface for nanoscale quantum mechanical simulations
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Leverage Schrödinger’s computational expertise and technology at scale to advance your projects through key stages in the drug discovery process.
Access expert support, educational materials, and training resources designed for both novice and experienced users.
