Active Learning Applications
Accelerate discovery with machine learning

Accelerate discovery with machine learning
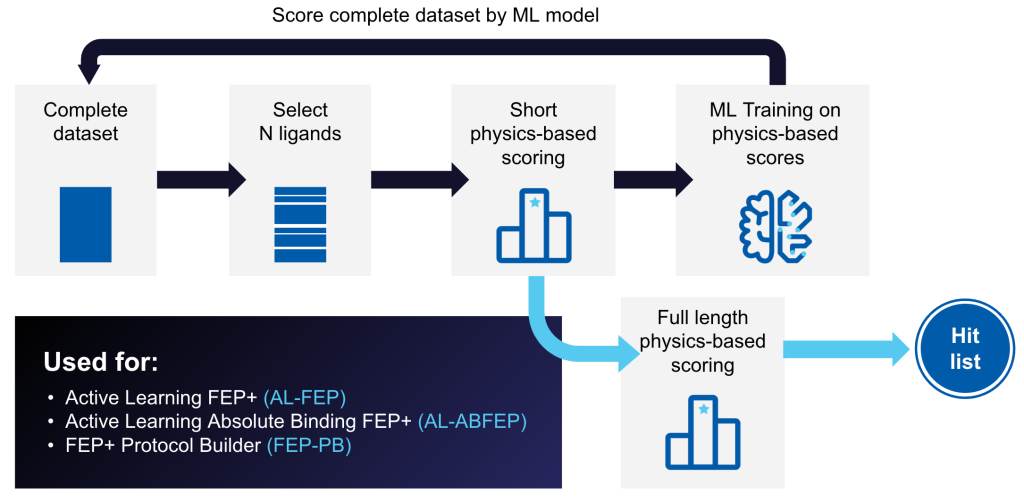
Active Learning Applications is a powerful tool that trains a machine learning (ML) model on physics-based data, such as FEP+ predicted affinities or Glide docking scores, iteratively sampled from a full library.
Trained models can rapidly generate predictions for new molecules and identify the highest-scoring compounds in ultra-large libraries at a fraction of the cost and speed of brute force methods.
Screen billions of compounds with Glide docking amplified by cutting-edge machine learning models in a fraction of the time. Use Active Learning to recover ~70% of the same top-scoring hits that would have been found from exhaustive docking of ultra-large libraries with Glide, for only 0.1% of the cost.
Explore tens of thousands to hundred of thousands of idea compounds with Active Learning FEP+, against multiple hypotheses simultaneously, to quickly identify compounds that maintain or improve potency while achieving other design objectives.
Rapidly generate accurate FEP+ protocols for systems that do not perform well with default settings. FEP+ Protocol Builder uses an Active Learning workflow to iteratively search the protocol parameter space to develop accurate FEP+ protocols, saving researcher time and increases the chances of successfully enabling FEP+.
Learn more
For Compounds
Enter in the numbers for your project (type in the box or use slider) to compare compute time and cost.
*Estimated customer compute costs only, based on $0.06 per CPU hour and $.35 per GPU hour. Recommended hardware for AL-Learning Glide.
License costs are not included. Contact us for a quote.
We assume 1M of the best ligands are docked with the final model.
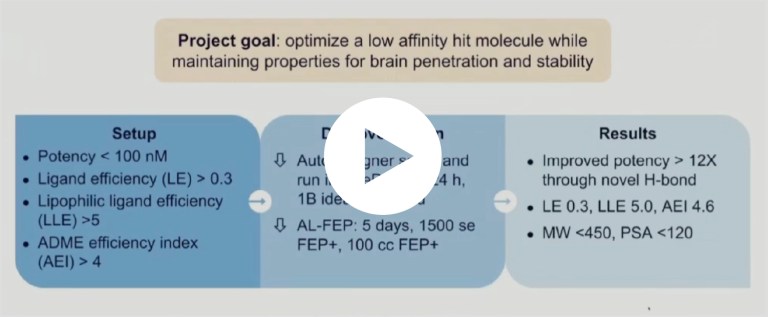
Schrödinger’s De Novo Design Workflow is a fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement.
Starting from a hit molecule or lead series, the technology identifies synthetically tractable molecules that meet key project criteria by combining multiple compound enumeration strategies with an advanced filtering cascade (AutoDesigner) and rigorous potency scoring with free energy calculations (Active Learning FEP+).

Get answers to common questions and learn best practices for using Schrödinger’s software.
Learn more about the related computational technologies available to progress your research projects.
Fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement

Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.