PrimeX
A comprehensive package for accurate protein crystal structure refinement

A comprehensive package for accurate protein crystal structure refinement
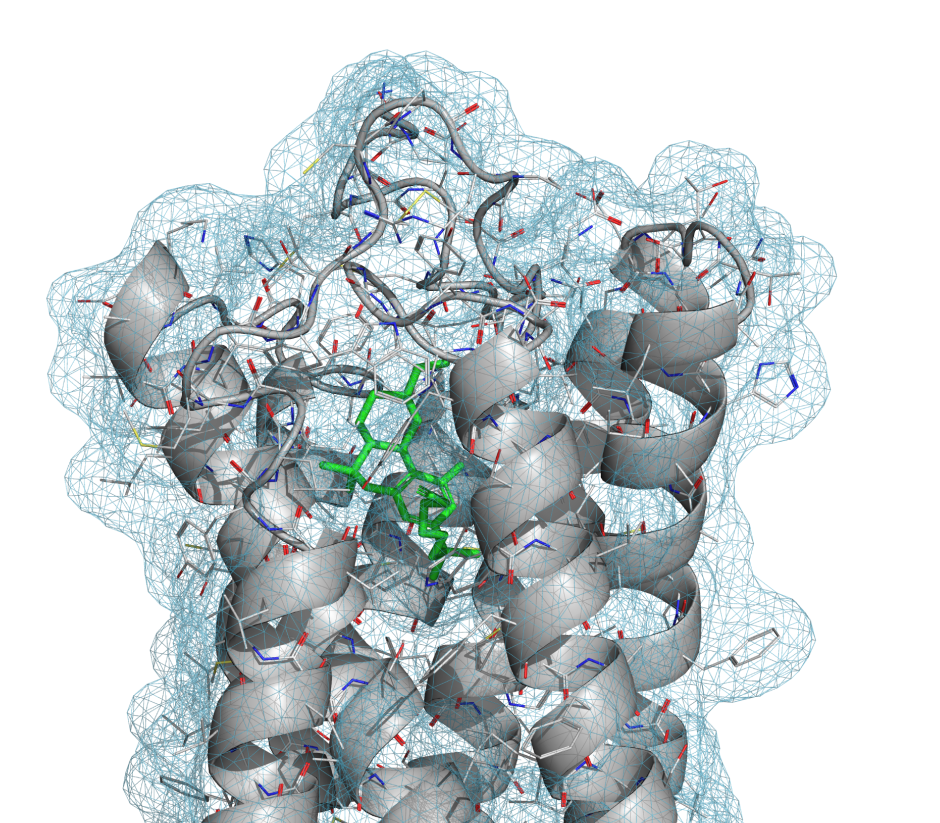
PrimeX is an advanced tool to facilitate protein crystal structure refinement and produces accurate structures more compatible with computational chemistry applications than those produced by other protein refinement programs.
Traditional protein crystallography introduces issues for downstream modeling due to high-energy contacts and missing hydrogens. PrimeX addresses these concerns by restraining protein geometry to OPLS-AA, adding hydrogens during refinement, and improving non-bonded interactions for better structure accuracy.
Builds loops up to 40-residues in length, using technologies in the well-validated Prime protein modeling program and guided by electron density fit.
Places ligands and other small molecules into electron density using technologies in the Glide docking program, which has demonstrated superior accuracy in ligand-receptor docking.
Utilizes the OPLS-AA force field with state-of-the-art computational technologies to refine protein structures that are immediately ready for all computational simulations.
Provides simulated annealing for reciprocal space refinement.
Offers conjugate gradient, truncated Newton, and quasi-Newton (LBFGS) to optimize performance and accuracy.
Generates parameters for ligands and other small molecules, as well as modified residues, automatically without requiring user intervention.
Automatically adds hydrogens, which are included during refinement according to physical chemistry as prescribed by the OPLS-AA force field.
Intuitive user interface is integrated into Maestro with step-by-step organization of refinement statistics in the Project Table and convenient analysis of protein structure geometry through interactive tables and plots.
Allows command-line input as well as scripting with Python for added control and customizable operations.
Get answers to common questions and learn best practices for using Schrödinger’s software.

Learn more about the related computational technologies available to progress your research projects.
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.