Machine Learning for Formulations Containing Proteins
Integrating AI and Machine Learning to Accelerate Composite Resin Formulation

MAY 13, 2026
Integrating AI and Machine Learning to Accelerate Composite Resin Formulation
Schrödinger is excited to be hosting a webinar in collaboration with Composites World, taking place on May 13th at 11:00AM EDT.
Artificial intelligence and machine learning have entered into everyday usage, but what impact can they have on polymer and ceramic matrix composites development?
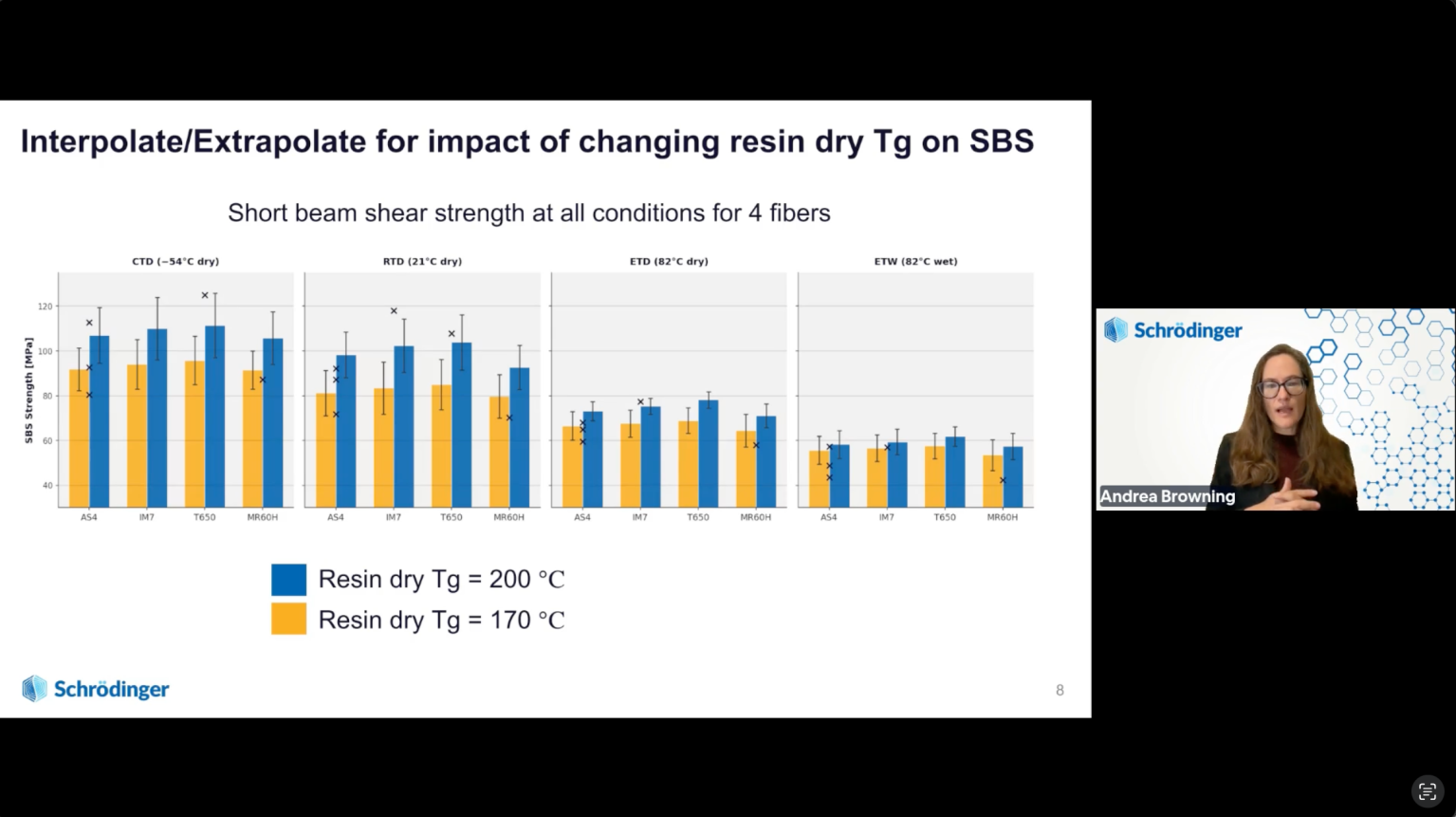
Composite performance depends heavily on matrix properties that govern processability and operational stability. Increased digitization is providing clear value across industries, but successful application in composite resin formulations requires a clear understanding of the key questions and insight into the critical design factors. Combining expert know-how and atomic-level detail with powerful artificial intelligence and machine learning tools enables resin formulation teams to maximize successful design initiatives.
This webinar will demonstrate how integrating machine learning with molecular simulation enables faster, more informed development of next-generation resin formulations.
Agenda:
- Where AI and machine learning add value: Discover how these technologies aid in designing polymer and ceramic matrix composites, focusing on critical matrix properties.
- Digitization: Learn why successful resin formulation requires increased digitization for both experimentation and simulation.
- Integration: See how combining chemistry expertise with AI and machine learning tools leads to better decision-making and outcomes.
- Acceleration: Explore how machine learning and molecular simulation accelerate the development of new resin formulations.
Our Speaker

Andrea Browning
Senior Director of Polymers and Soft Matter, Schrödinger
Andrea Browning, senior director of polymers and soft matter at Schrödinger, leads initiatives in polymer and soft matter simulations. Before joining Schrödinger, Browning was a lead research engineer and project manager at Boeing, where she focused on translating engineering problems into fundamental materials insights. She brings more than a decade of experience in connecting industrial and engineering problems to root materials issues and how simulations can be used to inform industrial decisions. Browning earned her doctorate in chemical engineering from the University of California, Santa Barbara, where she was a National Science Foundation Graduate Research Fellow.
Large-scale Atomistic Simulations of Lithium Diffusion in a Graphite Anode with a Machine Learning Force Field
Efficient Long-Range Machine Learning Force Fields for Liquid and Materials Properties
Advancing efficiency in deep-blue OLEDs: Exploring a machine learning–driven multiresonance TADF molecular design
Band Gap and Reorganization Energy Prediction of Conducting Polymers by the Integration of Machine Learning and Density Functional Theory
Screening Antioxidant Ingredients Using Quantum Mechanics and Machine Learning
Accurate hydration free energy calculations for diverse organic molecules with a machine learning force field
Advancing battery materials innovation using charge-aware machine learning force fields

OCT 29, 2025
Advancing battery materials innovation using charge-aware machine learning force fields
Batteries are fundamental technology – powering everything from our personal electronics to electric vehicles, as well as large-scale grid storage systems for renewable energy integration. However, current battery technologies, primarily lithium-ion batteries, face significant limitations in performance, safety, cost, and reliance on scarce materials like cobalt. Therefore, innovation in battery materials is the key to unlocking the next generation of energy storage.
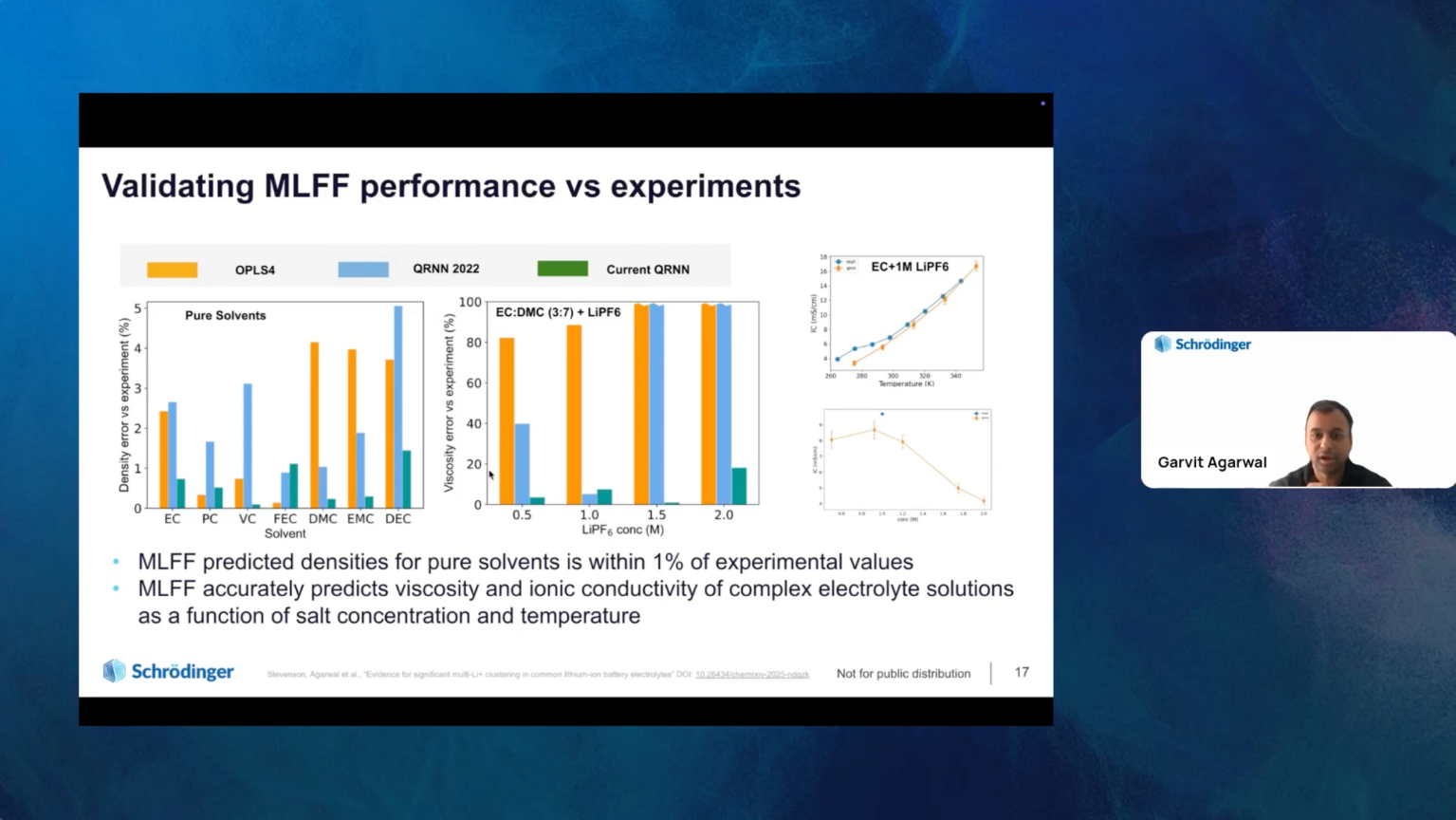
In this webinar, we will demonstrate how Schrödinger is utilizing an integrated computational approach combining physics-based molecular modeling with machine learning force fields (MLFFs) to address key challenges in battery materials design. We will introduce Schrödinger’s latest advancements in MLFFs, featuring charge recursive neural networks (QRNN) and the recently released Message Passing Network with Iterative Charge Equilibration (MPNICE) architectures, which incorporate explicit electrostatics for accurate charge representations.
Moreover, we will showcase several industry-relevant case studies highlighting the application of MLFFs to precisely model the structure and properties of electrolyte materials (liquid, polymer, and inorganic solid-state electrolytes), cathode coatings, and electrode materials. We will also explore how MLFFs facilitate large-scale simulations, allowing scientists to investigate the impact of defects and heterogeneities on crucial properties like Li-ion transport, paving the way for the efficient design of next-generation battery materials and chemistries.
Webinar Highlights:
- How Schrödinger combines physics-based modeling with machine learning force fields to drive battery materials discovery
- Schrödinger’s latest MLFF technologies, including QRNN and MPNICE
- Real-world case studies modeling electrolytes, cathode coatings, and electrode materials
- How MLFFs facilitate large-scale simulations, such as the investigation of Li-ion transport
Our Speaker

Garvit Agarwal
Principal Scientist, Schrödinger
Garvit Agarwal, Principal Scientist and Scientific Lead for Energy Storage at Schrödinger, works to extend and apply molecular modeling tools for the accelerated discovery of next-generation clean energy technologies. Garvit obtained his Ph.D. in Materials Science and Engineering from the University of Connecticut. He worked as a post-doctoral researcher in the Materials Science Division at Argonne National Laboratory prior to joining the Materials Science team at Schrödinger.
Machine Learning Force Fields
Advancing machine learning force fields for materials science applications 最新機能 MPNICEのご紹介

Advancing machine learning force fields for materials science applications
最新機能 MPNICEのご紹介
機械学習力場(MLFFs:Machine Learning Force Fields)は、「機械学習原子間ポテンシャル」とも呼ばれ、多様な化学系に対するコスト効率の高い原子レベルのシミュレーションを実現するための重要なツールとして登場しており、しばしば密度汎関数理論(DFT)に匹敵する精度を、はるかに低い計算コストで達成しています。
近年のメッセージパッシングネットワークの進歩により、従来のMLFFが抱えていた「対応できる元素の種類に制限がある」という課題が克服されました。さらに、電荷平衡法を用いた原子電荷および静電相互作用の導入により、複数の電荷状態、イオン系、電子応答特性の精密な再現が可能となり、長距離相互作用を明示的に考慮することで、さらに高い精度を実現しています。
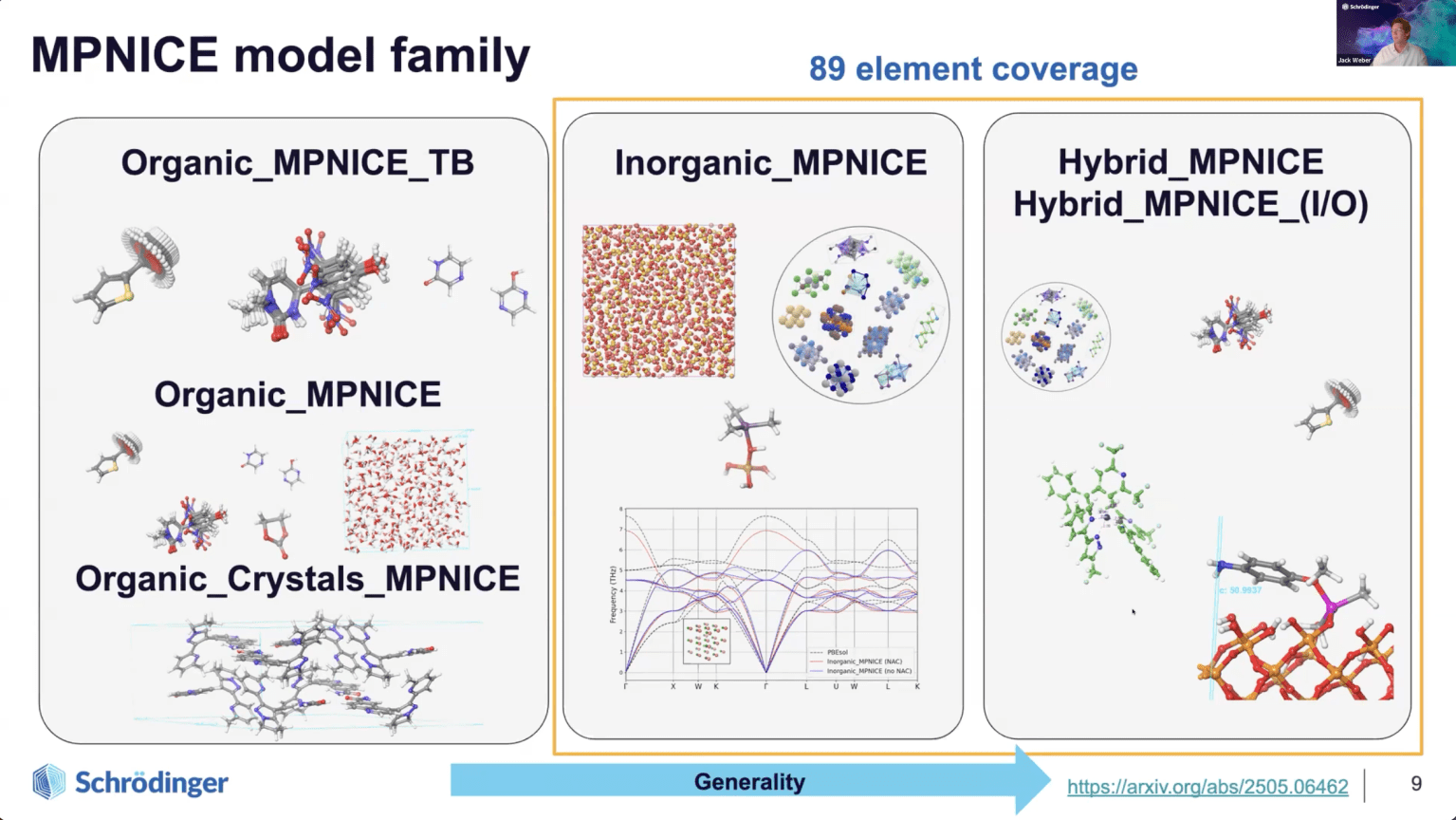
本ウェビナーでは、シュレーディンガーが開発した最先端のMLFFアーキテクチャ「MPNICE(Message Passing Network with Iterative Charge Equilibration)」をご紹介します。MPNICEは、正確な電荷表現のために明示的な静電気を組み込んでいます。周期表全体(89元素)を網羅する材料を対象に学習させた事前学習済みモデル群も提供しています。
MPNICEは高いスループット性能を重視しており、従来の手法では実現困難だった長時間・大規模な原子レベルのシミュレーションを、高精度を維持しながら可能にします。
ウェビナーでは、材料設計においてより大規模かつ複雑なシミュレーションを可能にするMLFF搭載ツール群の概要を紹介し、産業応用に即した事例を交えて解説します。
ハイライト:
- MPNICEの概要:原子の部分電荷や長距離相互作用を取り入れながら、同等精度のモデルよりも1桁高速な計算を実現する、メッセージパッシング型機械学習力場(MLFF)アーキテクチャ
- MPNICEの最新応用例の紹介:産業界のニーズに対応するために、有機材料、無機材料、そして有機・無機ハイブリッド材料に対する汎用モデルとして活用された事例を紹介
Our Speaker

Jack Weber
Senior Scientist, Schrödinger
コロンビア大学で化学物理学の博士号を取得。博士課程では、先端的な計算手法を駆使し、化学および材料科学における基礎的課題に取り組み、遷移金属錯体のような強相関系に対応するための電子構造計算手法(ab initio法)の改良や、三重項-三重項消滅(TTA)型アップコンバージョン光触媒の設計などを行いました。 現在は、創薬および材料科学分野における応用を目的とした機械学習力場(MLFF)の開発に取り組んでいます。