Welcome to the Schrödinger Maestro workshop at CSC! This year’s workshop focuses on small molecule drug discovery and drug formulation with Schrödinger Maestro software suite. No prior simulation experience is required. The focus will be on hands-on experience and application.
It’s possible to participate on-site at CSC premises in Espoo, or join via Zoom (link provided for registered participants). Registration is required, but the event is free of charge both onsite and online.
Speakers:
Irene Bechis, Senior Scientist I, Schrödinger
Philipp Dohmen, Senior Scientist I, Schrödinger
Day 1: Wednesday, December 4th 2024
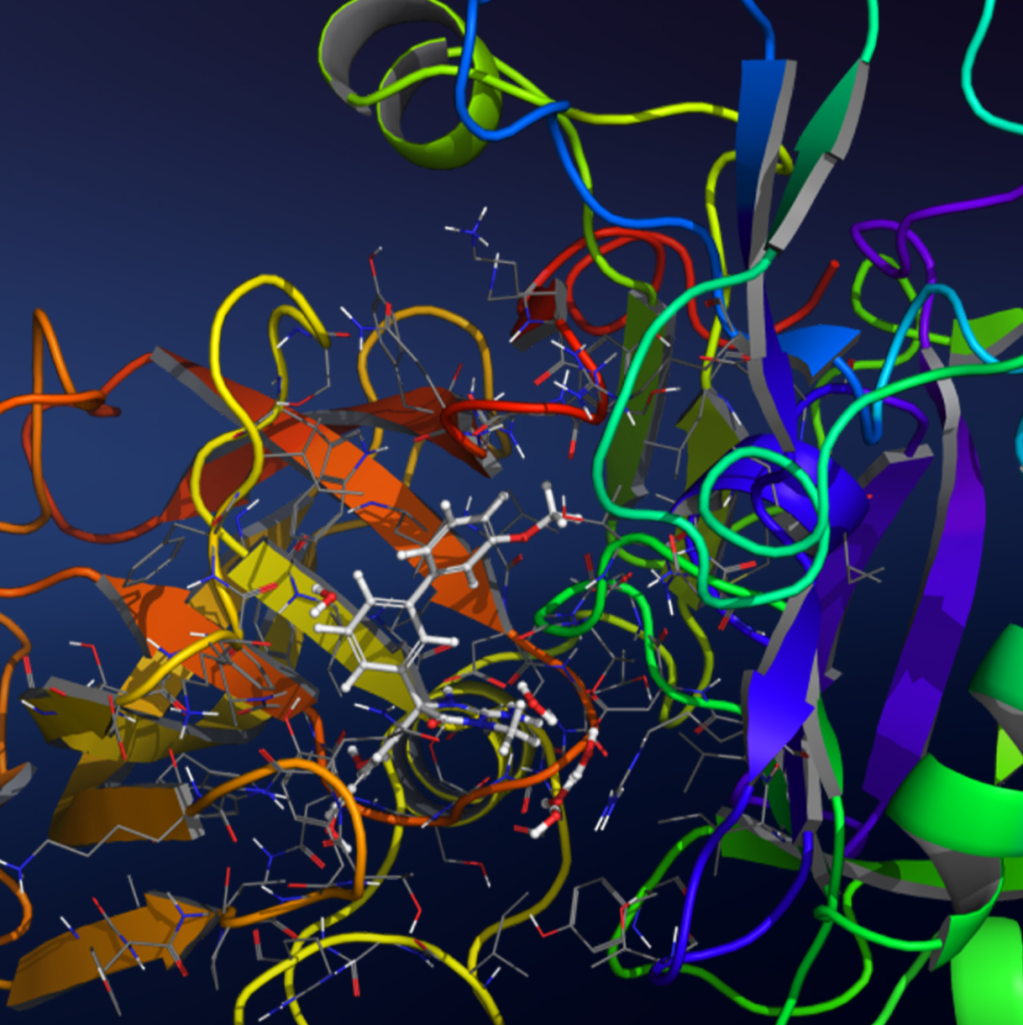
Small molecule drug discovery part I: Computational modeling can support and enhance various stages of the drug discovery and design process. The Maestro interface contains all the tools needed to import and prepare your starting small molecule and protein system, and provides access to the computational tools within the Schrödinger suite for life sciences. In this session we will go through the necessary steps to prepare ligand and protein structures. We will then explore ligand design in an automated fashion using the Ligand Designer GUI, which facilitates on-the-fly ideation through ‘build and dock’ workflows. Finally, we will set up and run docking calculations with Glide, and analyze how the resulting docked compounds satisfy the basic criteria of shape and molecular interactions that lead to the final Glide scoring term.
Drug formulation part I: A smart, strategic drug formulation can efficiently advance your drug development projects and inform downstream processes. Simulations can help selecting and combining the right formulation ingredients in the appropriate manner. In this session, we will introduce our Materials Science Suite for materials science applications, demonstrating the tools available for pharmaceutical formulations and drug development processes. We will guide you through building systems, running molecular dynamics simulations, and calculating the miscibility of active pharmaceutical ingredients (APIs).
Day 2: Thursday, December 5th 2024
Drug formulation part II: In this session we will focus on extracting key properties and information from simulations of various formulations. Prediction of chemical and physical stability of formulations in storage conditions are pivotal for understanding a product’s shelf life. In particular, prediction of the glass transition temperature (Tg) for APIs and mixtures is an important indicator of thermodynamic stability in the solid state and a relevant information for the
manufacturing process. Similarly, predicting the tendency for a formulation to uptake water at certain humidity conditions is important for knowing its behaviour and stability in different storage conditions. Using MS Maestro and the tools available in the MS Suite, we will calculate Tg and hygroscopicity of APIs and API/polymer mixtures. Finally, we will introduce Coarse Grained simulations, useful for describing those complex and evolving structures, often in fluid states, that play a crucial role for API delivery.
Small molecule drug discovery part II: By studying the dynamics of a molecular system in a thermodynamic environment, we can investigate processes such as protein folding, protein-protein or protein-ligand interactions, and gain new structural insights for drug discovery and design. In this session we will learn how to set up, run and analyze the results of an unrestrained all-atom molecular dynamics simulation using Desmond and Maestro. We will focus on a protein-ligand complex and go through different ways and tools to analyze a simulation. The analysis will improve our understanding of the binding pocket and the interactions between protein and ligand.